Illumina MiSeq Dual-barcoded Two-step PCR Amplicon Sequencing Protocol
Jana M. U'Ren, A. Elizabeth Arnold
Abstract
Background Information
- High throughput amplicon sequencing is an extremely sensitive technique. The lab environment is full of airborne bacterial and fungal cells as well as bacterial and fungal genomic DNA from cultures, and so extreme care must be taken to keep reactions uncontaminated and in preventing cross-contamination.
- This protocol was modified from the IBEST Geomics Core at University of Idaho. It utilizes a 2-step PCR, where the first PCR amplifies the specific region of interest (e.g., ITS or 16S V4), and the second PCR adds adapters and barcodes to these amplicons. The target specific primers include (in addition to the standard primer sequence) the universal sequences CS1 and CS2, which provide the bases used in the second PCR to extend the amplicons with Illumina adapters and sample-identification barcodes.
- Illumina adapters and sample-identification barcodes are not included in the region-specific primers used in PCR1. This allows for maximum flexibility in choosing which primers to use and provides the ability to swap out targets, or include multiple targets in the same sequencing reaction, without needing to purchase a large number of barcoded target specific primers.
- Barcodes are included in both adapters, therefor a pair of barcodes is used to uniquely identify each sample during bioinformatic processing. This allows us to use 32 barcode pairs to uniquely identify 1024 samples (32 x 32).
- Phase-shifted primers are used to introduce 1, 2, and/or 3 additional bases to allow for the maximum amount of “sequence diversity” in the first 4 bases. This is important because of how the Illumina sequencers detect clusters on the flowcell during sequencing. If sequences are too similar (as is often the case with amplicons), then sequencing yields will be much lower than expected. 2-bp linkers and phase-shifting bases were chosen to have low identity between primers and the target sequence.
- Tagged primers were ordered from IDT (standard desalting; not HPLC-purified), re-suspended at 100 μM using a new bottle of molecular grade water in the PCR-free hood, and mixed in equimolar concentration (250 μl each). Working stocks were diluted to 50 μM for PCR1.
Before start
Steps
DNA QUANTIFICATION
For best results, first quantify DNA extractions using a PicoGreen assay with a fluorometer (e.g., Qubit). If possible, normalizing all DNA pools to a standard level (e.g., 5-10 ng/ul) prior to PCR1 ensures more consistent amplification across samples.
For soils, IBEST recommends using 100 ng of DNA for the first PCR step. However, when working with samples containing PCR inhibitors, or samples with low sample volume or low concentration, this may not be possible. Try to use between 10-100 ng of DNA per reaction. In general, use as much DNA as you can without causing PCR inhibition.
PCR1 using Phusion Flash High-Fidelity PCR Master Mix
Phusion Flash MM is a high-fidelity, hot start enzyme that enables extremely quick PCR runs. It is based on the polymerase from Pyrococcus furiosus instead of Taq, which comes from the polymerase in Thermus aquaticus .
It is much higher fidelity (which means fewer errors), something that is important because of the high sensitivity of high throughput amplicon sequencing. In my experience, it works better than other enzymes, but this may be dependent on the sample type, thermocycler, etc. that is being used.
Be aware that the products that result from PCR1 must contain only very minimal primer dimers for optimal sequencing results. This can be accomplished if care is taken to minimize primer concentrations in the reaction. If primer dimer is an issue, you will need to optimize your PCR1 reaction using a lower primer concentration (IBEST recommends a final concentration of 0.05 μ M). Obtaining clean and correctly sized PCR1 products is critical to ensure that barcodes will properly attach during PCR2.
If using mPNA/pPNA to block amplification of host mitochondrial and plastid SSU sequences (for 16S PCR only), decrease the amount of water in each reaction and add 0.4 µl of each 50 µM PNA (1.25 µM final concentration).
All PCRs should include a positive template and negative template controls (PTC and NTC, respectively). All PCRs should be run in triplicate (3 times for each sample) to reduce the stochasticity associated with PCR amplification of diverse microbial communities.
| A | B | C |
|---|---|---|
| PCR1 Mastermix (1 reaction) | volume | final conc. |
| Phusion Flash MM (ThermoFisher F-548L) | 7.5 µl 1X | |
| 50 µM CS forward primer | 0.80 µl | 0.5 µM * |
| 50 µM CS reverse primer | 0.80 µl | 0.5 µM* |
| BSA 20mg/ml (NEB cat# B9000S)* | 0.80 µl | 1.0 mg/ml* |
| Nuclease-free PCR grade water | 4.30 µl | |
| Template DNA | 1.0µl | ~10-100 ng/µl* |
| Total reaction volume | 15 µl |
| A |
|---|
| ITS/V4 PCR1 conditions (Phusion MM)* |
| 98°C for 10 sec |
| 98°C for 1 sec |
| 57°C for 5 sec |
| 72°C for 20 sec |
| Go to 2 25X |
| 72°C for 1 minute |
| 4°C forever |
*Please be aware these concentrations and thermocycling conditions are only a suggested starting point . You may need to optimize to ensure the highest quality PCR1 products.
2% agarose gel analysis of PCR1 products
Prepare a new 2% gel with new TAE buffer before each gel. Our lab uses Use SYBR green, but ethidium bromide or Gel Red can also be used to view products on a UV transilluminator. Do not reuse old gels or buffer! These reagents are expensive, and the technique is very sensitive, so we want to be confident in our gel images.
Centrifuge PCR1 products for 1 minute at maximum speed in a 96-well plate centrifuge.
On the gel bench, mix 2 µl of SYBR and 2 µl of loading dye for each reaction in a clean 96-well plate.
In the UV treated and DNA Away cleaned PCR cabinet, mix 3 µl of the PCR1 product with loading dye and SYBR mixture. Gently pipette to mix.
Gentle pipetting reduces the formation of aerosols or bubbles, which could easily cross-contaminate the samples.
After this step, the original PCR1 products can be stored at 4ºC for up to 3 days or at -20ºC for longer periods.
Load this mixture onto the 2% agarose gel. Run until ladder is sufficiently separated to size product accurately, generally around 30 minutes.
See Fig 1 for an example PCR1 gel image from IBEST. Ideally, PCR1 products have relatively faint (due to a lower number of cycles), but distinct bands and very little to no primer artifact (primer diner). (Image from IBEST)
Fig. 1

Prepare the dilution of the pooled PCR1 products
Once acceptable amplification is confirmed on an agarose gel for each of your reactions, pool triplicate PCRs for each sample in the sterilized (UV, DNA-away) PCR cabinet.
In a decomtaminated PCR cabinet, prepare a suitable dilution of the pooled PCR1 products. For example, we found a 15-fold dilution of PCR1 amplicons performed the best for most samples. To do so, add 5 µl of PCR1 product to 70 µl of PCR grade water, mix gently by pipetting, then centrifuge briefly for 30 seconds to spin down all components.
*If PCR1 amplification was faint, PCR1 amplicons can be diluted less (e.g., a 1:5 dilution: 5 µl of PCR1 product to 20 µl of PCR grade water) to ensure sufficient template for PCR2.
**Please be aware these dilutions are only a suggested starting point . You may need to optimize to ensure the highest quality PCR2 products.
PCR2 using Phusion Flash High-Fidelity PCR Master Mix
The purpose of PCR2 is to add adaptors to the PCR1 products, using between 5-10 additional cycles of PCR.
Barcoded primers are diluted to a concentration of 2 µM. Each well of the 96-well primer plate contains the F and R primer for PCR2. EXTREME CARE MUST BE TAKEN NOT TO CONTAMINATE PRIMER STOCKS!
Make a PCR2 mastermix containing Phusion, BSA, and water in the pre-PCR hood. Aliquot 18.25 µl for each reaction into strip tubes or a 96-well plate on the ice block.
PCR2 Mastermix (1 reaction) volume final conc.
Phusion Flash MM (ThermoFisher F-548L) = 10.0 µl 1X
2 uM Barcoded primers* = 0.75 µl 0.075 µM
BSA 20mg/ml (NEB cat# B9000S) = 0.24 µl 0.24 mg/ml*
Nuclease-free PCR grade water = 8.01 µl
Diluted PCR1 products = 1.0µl
Total reaction volume = 20 µl
*Please be aware these concentrations and thermocycling conditions are only a suggested starting point . You may need to optimize to ensure the highest quality PCR2 products.
In the same hood, add 0.75 µl of barcoded primer from the 96-well plate of “working” PCR2 primer stocks using a multichannel pipette. Close caps on strip tubes or seal the plate with an aluminum lid.
Transfer the plate with mastermix to the (again) UV-sterilized PCR cabinet. Carefully add 1 µl of diluted PCR1 product to each well, mixing gently by pipetting. Seal the plate with strip cap tubes (not foil!) to prevent evaporation and run on the thermocycler.
| A |
|---|
| ITS/V4 PCR2 conditions (Phusion MM)* |
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
*Please be aware these concentrations and thermocycling conditions are only a suggested starting point . You may need to optimize to ensure the best quality PCR2 products.
PCR 2 should contain two negative template controls (NTCs). The first is a standard water NTC. The second NTC is mastermix + 1 uL of the NTC from PCR1. The former will tell you if your mastermix is contaminated and the latter will tell you whether your water used for dilution was contaminated with amplicons.
Quality analysis of PCR2 products
For a subset of your samples, you should check 1 µl of PCR product on an Agilent BioAnalyzer (BA) using the Agilent DNA 1000 kit. Follow the Agilent DNA 1000 kit Guide for details.
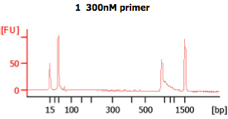
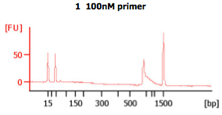
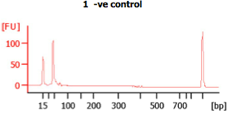
These BA traces (figures below) show that when excess concentrations of primers are used (i.e., when 300 nM of primer are used), large amounts of primer-dimers are present in the PCR product. In the same figure, a negative control (i.e., -ve control) is shown with these high primer concentrations. The peak seen next to the lower marker (at 35bp) results from primer dimers (Images from IBEST).
The next image shows a BioAnalyzer trace of a negative control (or NTC) and a sample (i.e., sample 3) after PCR2. The peaks at 15bp and 1,500bp are internal markers. (Images from IBEST)
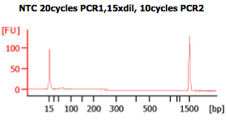
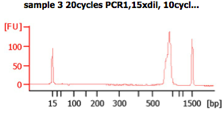
Check the BioAnalyzer results to determine if the PCR product has the expected size. PCR2 products should exhibit a band shift of ~59 bp when compared to first step PCR products (this represents the addition of the Illumina sequencing primers and barcodes).
Since it is not practical or cost-effective to run all samples on a chip, a 2% agarose gel may be run for the remaining PCR2 products to confirm high quality amplification (see above for details).
Store the PCR2 products at -20ºC.
PCR2 quantification and normalization
Quantify final PCR products with a fluorometer using PicoGreen and suitable standards. This can be done for a small number of samples on the Qubit using the Broad Range kit (i.e., BR kit), but, for larger number of samples, it is more cost-effective to quantify samples in a 96-well format. This can be done using a 96 well plate reader.
If sending samples to the sequencing core for quantification, make a 1:5 dilution of the PCR2 products (i.e., 2 µl of PCR2 product in 8 µl of molecular grade water) in a new 96-well plate. Mix gently by pipetting and seal well with a foil lid. Submit to the core for quantification.
Normalize PCR2 products to a set concentration (e.g., 5 ng/µl) by adding different amounts of molecular grade water to each well of a 96-well plate. Using a multichannel pipette, transfer a set amount of PCR2 product to each well (i.e., 4 µl). These amounts will vary based on how well your extractions and amplifications worked, and you will have to calculate the amount of water needed to dilute each sample based on the quantifications you have performed (above).
Library pooling
In the decomtaminated post-PCR hood, pool between 20-100 ng of DNA for each sample (but the same amount for each sample) per run into a sterile 2 mL microcentrifuge tube.
**If pooling amplicons from loci that differ in size make separate pools for each locus, even if they are to be sequenced on the same run.
**In cases where critical samples had poor amplification, these can be submitted in a separate pool as “poor” samples. Pooling “poor” samples with the high quality amplicons brings down the quality of the entire library and should be avoided.
Clearly label tubes containing the various sample pools and extentively parafilm the lids of all tubes. This is essential because of low air pressure during air shipment, which can cause the lids of the tubes to pop open if not properly sealed and parafilmed.
To protect samples during shipment, store microcentrifuge tubes in a larger 50 mL Falcon tube and fill any remaining space in the tube with Kimwipes.
Freeze tubes at -20 for transport. Library preparation will be done at sequencing core.
Sample submission
Fill out sample submission sheet with sample name, primers, and barcode information.
