Norovirus genotyping and phylogeny analysis_ViroTrakr workflow 1_v.1
Zhihui Yang, Jayanthi Gangiredla, Mark Mammel
Disclaimer
Please note that this protocol is public domain, which supersedes the CC-BY license default used by protocols.io.
Abstract
This workflow provides step-by-step instructions for norovirus analysis within the GalaxyTrakr platform. It includes the quality assessment for raw sequencing data (from most next-generation sequencing platforms), drafting de novo assemblies, and reporting the sequence genotype and phylogenetic results. This workflow was designed for norovirus, which is one of the major targets of our ViroTrakr database.
This protocol covers how to:
Set up an account in Galaxy Trakr;
Create a new history/workspace for a new submission;
Upload raw data obtained from local folders or download from NCBI;
Execute the ViroTrakr workflow 1 (norovirus);
Interpret the results.
ViroTrakr:
foodborne viruses (ID 396739) - BioProject - NCBI (nih.gov)
Reference: Quality control assessment for microbial genomes: GalaxyTrakr MicroRunQC workflow V.5:
Quality control assessment for microbial genomes: GalaxyTrakr MicroRunQC workflow (protocols.io)
Steps
Log into your GalaxyTrakr account.
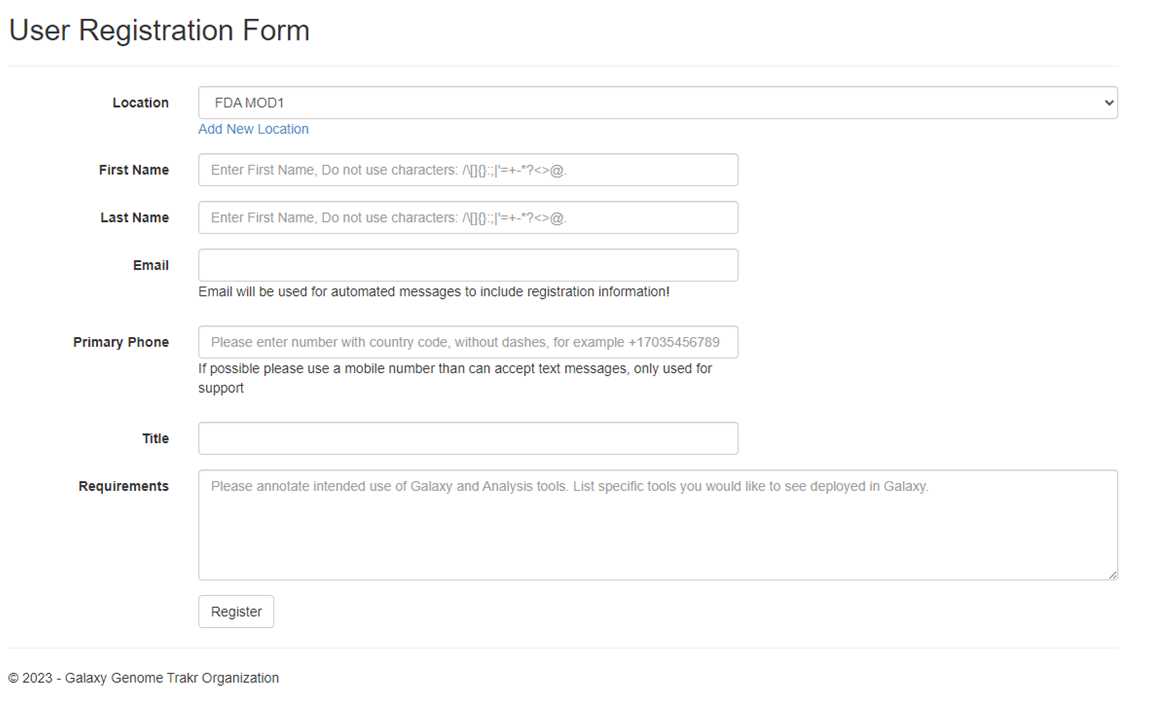
Create a GalaxyTrakr account if you are the first-time user:
User Registration Form - Galaxy Genome Trakr (galaxytrakr.org)
Log into your GalaxyTrakr account if you already have one:Galaxy (galaxytrakr.org)
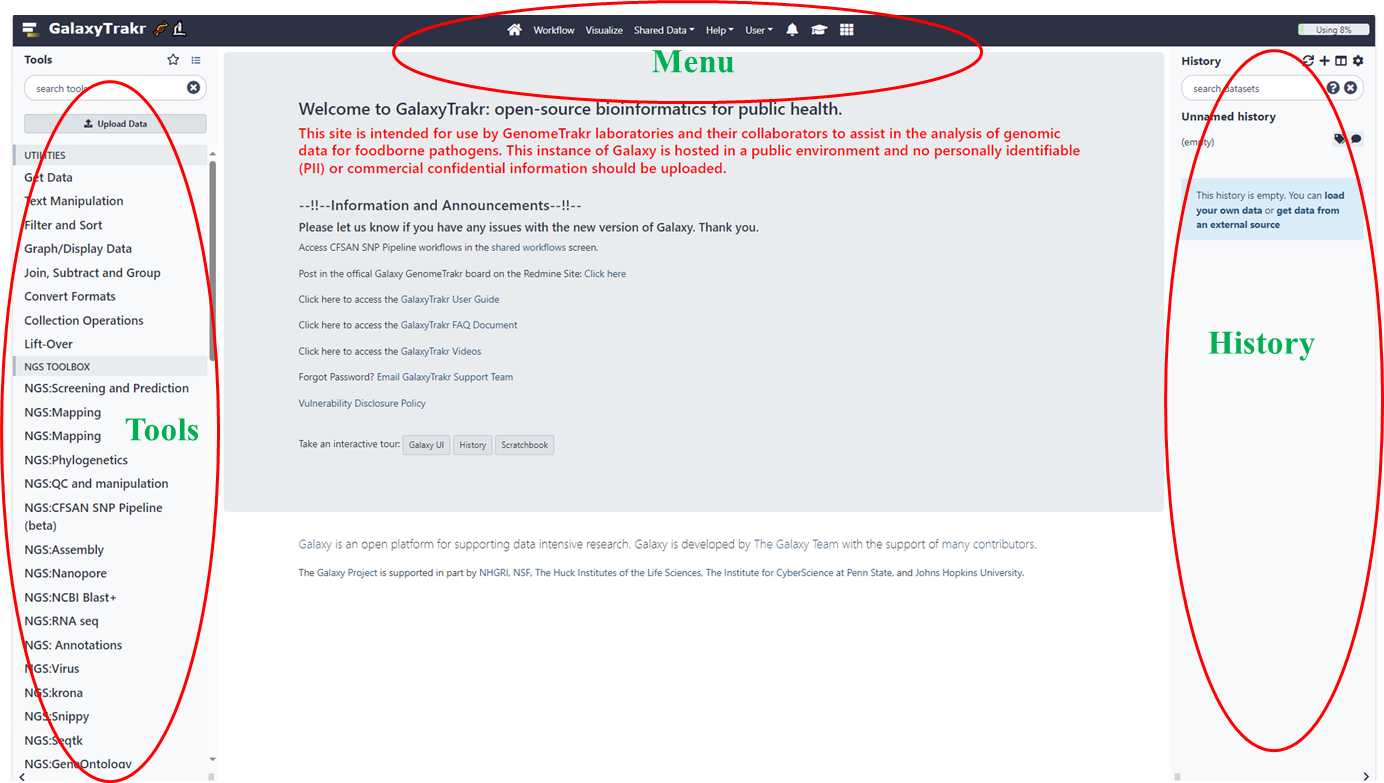
Get familiar with Galaxy components: Tools, Menu and History.
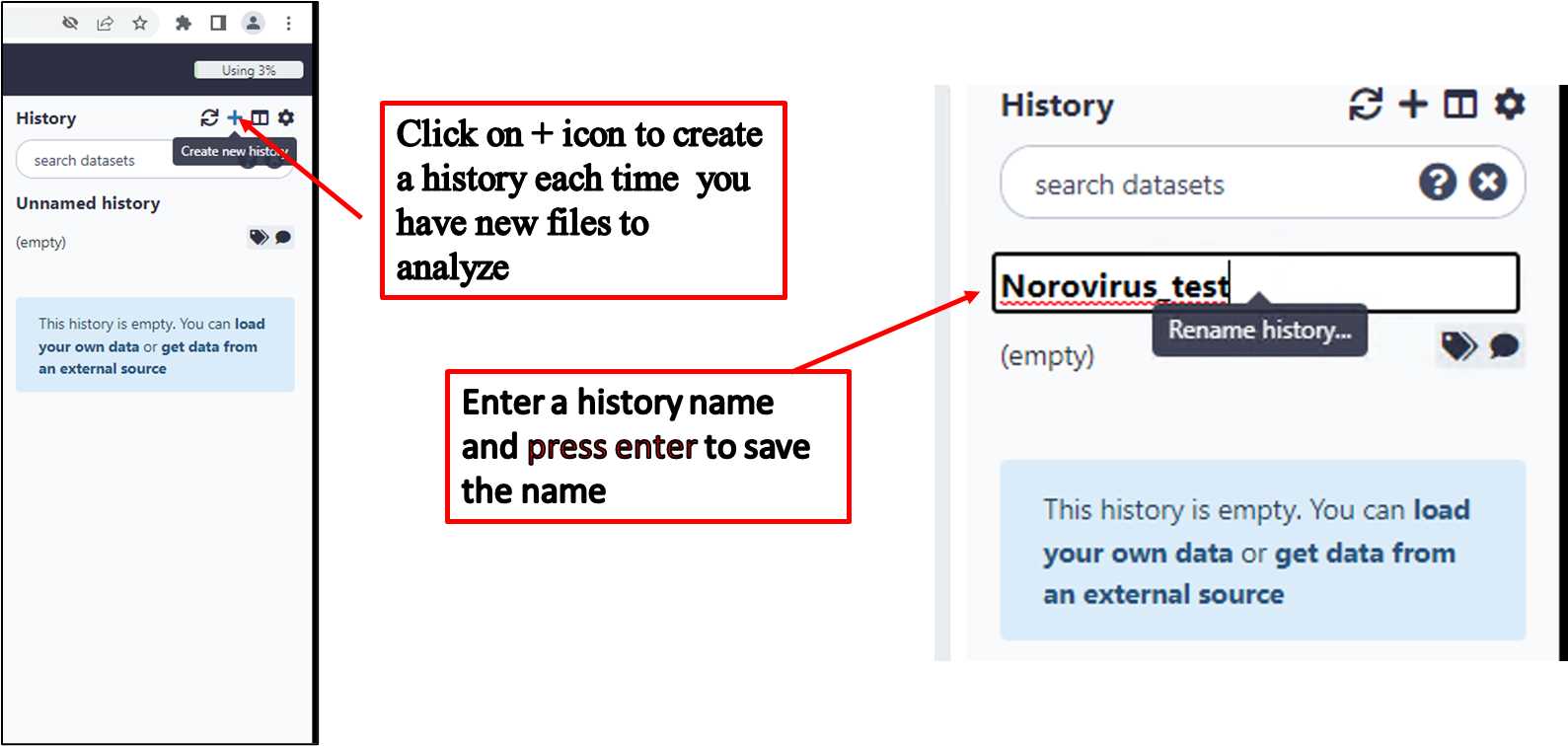
Create a new history.
Upload data.
The raw sequencing data in fastq files can be imported into GalaxyTrakr directly from your local folder (instructions shown in 3.1); or downloaded from SRA (instructions shown as in 3.2) if the files have been already submitted to ViroTrakr in NCBI (Submission protocol: NCBI submission protocol for foodborne virus surveillance (protocols.io)). After being uploaded to GalaxyTrakr, the files will remain in your account until they are deleted.
Upload raw data from local folder.
3.1.1. Click on the button “Upload Data”, then “Choose local files”.
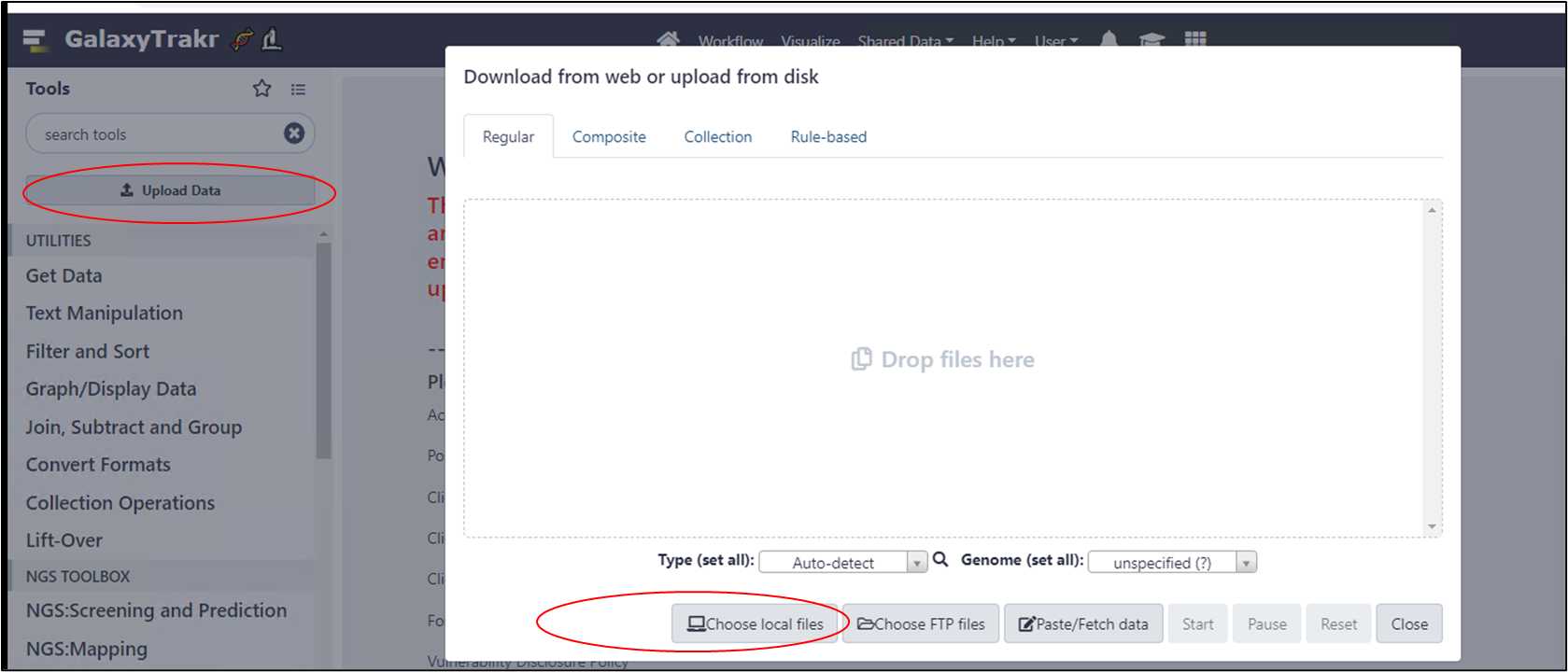
3.1.2. Select fastq files from your local folder.
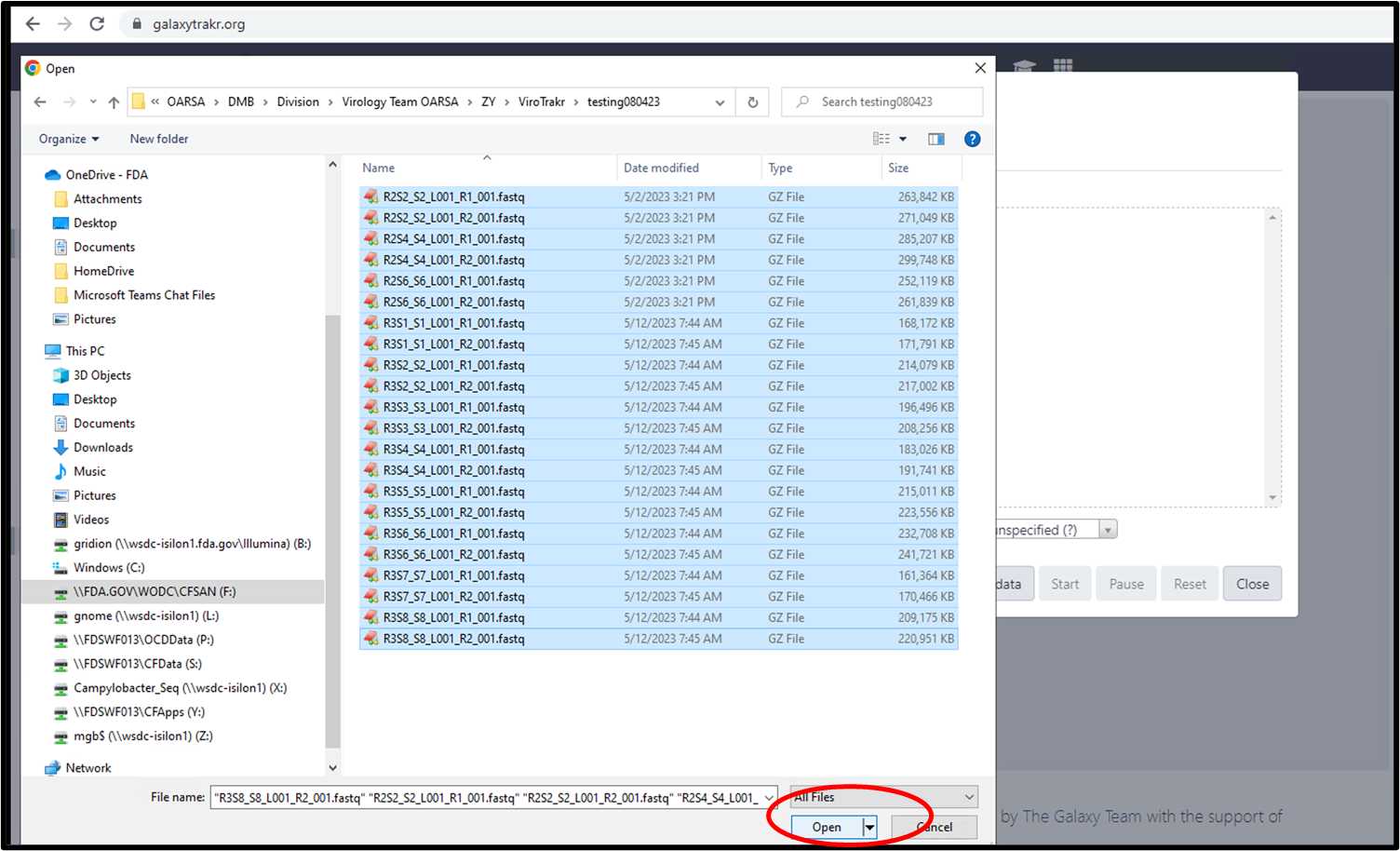
3.1.3. Select the files and click “Start” to upload.
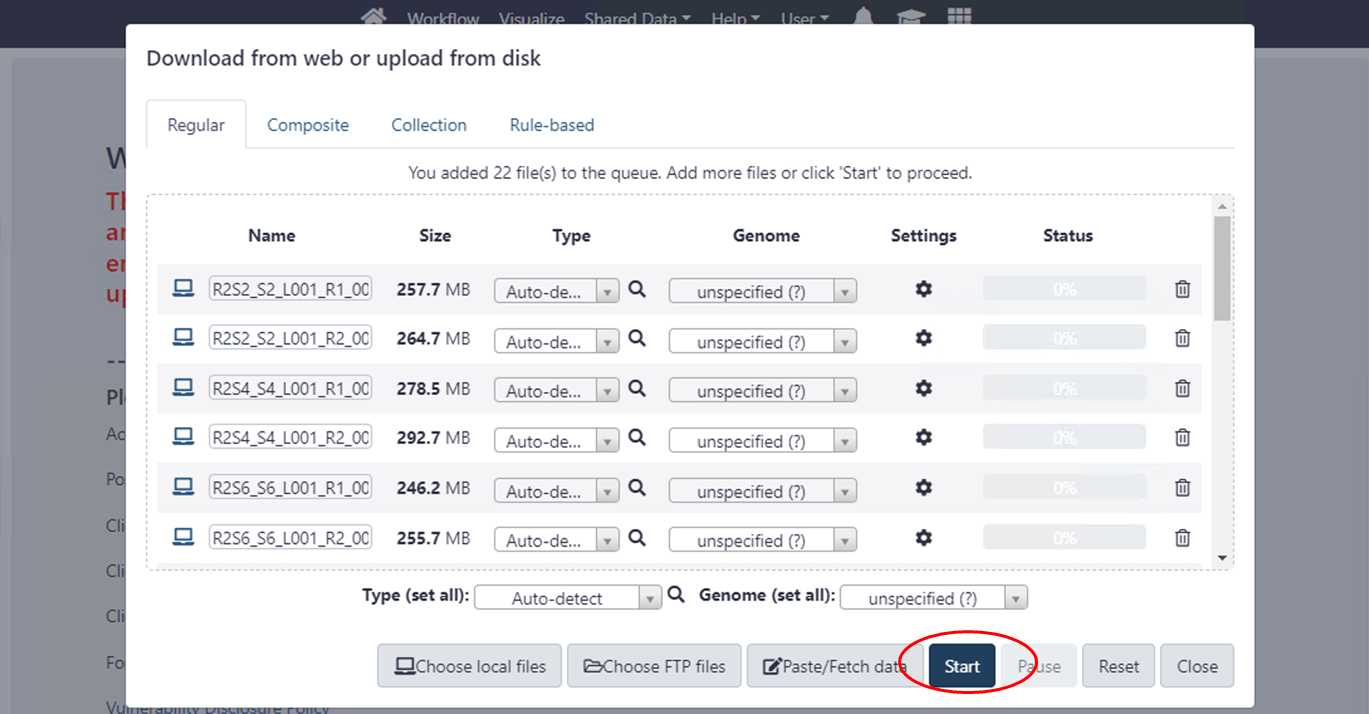
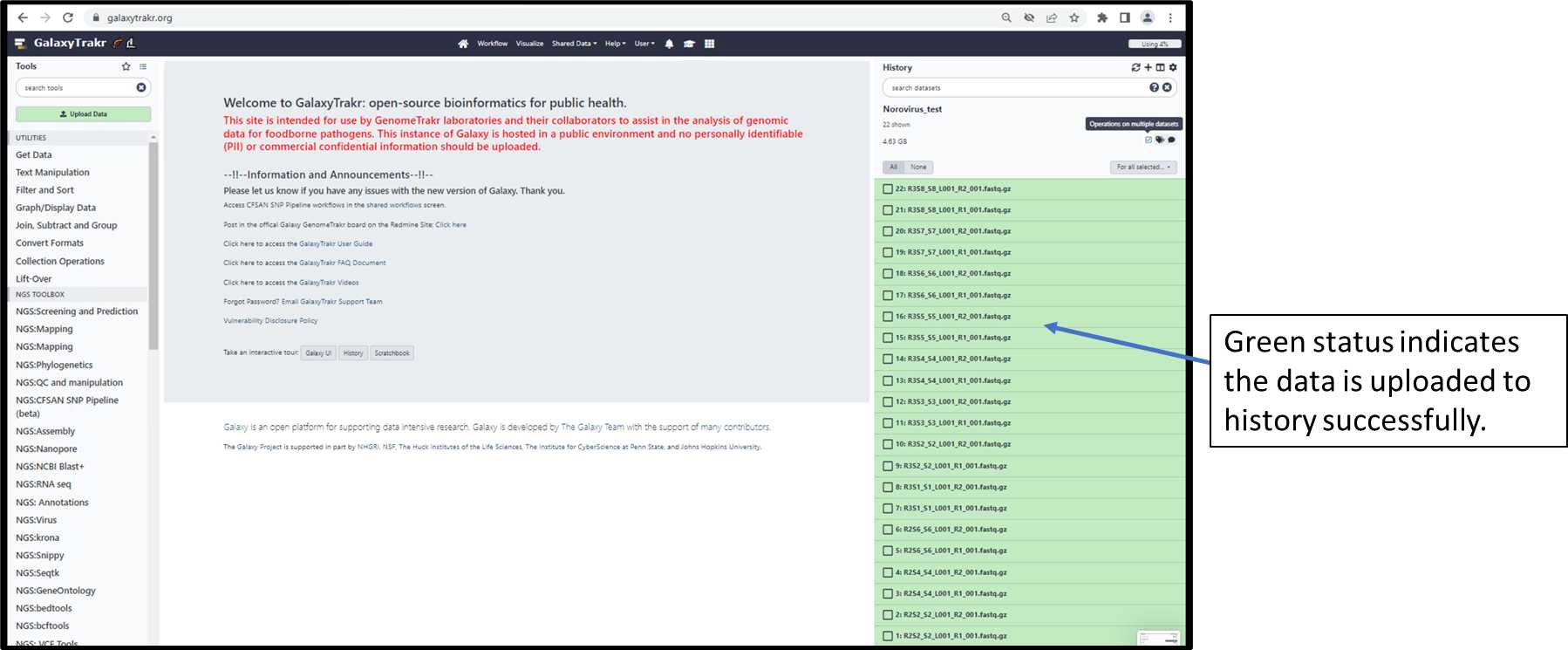
3.1.4. Check the status of data upload.
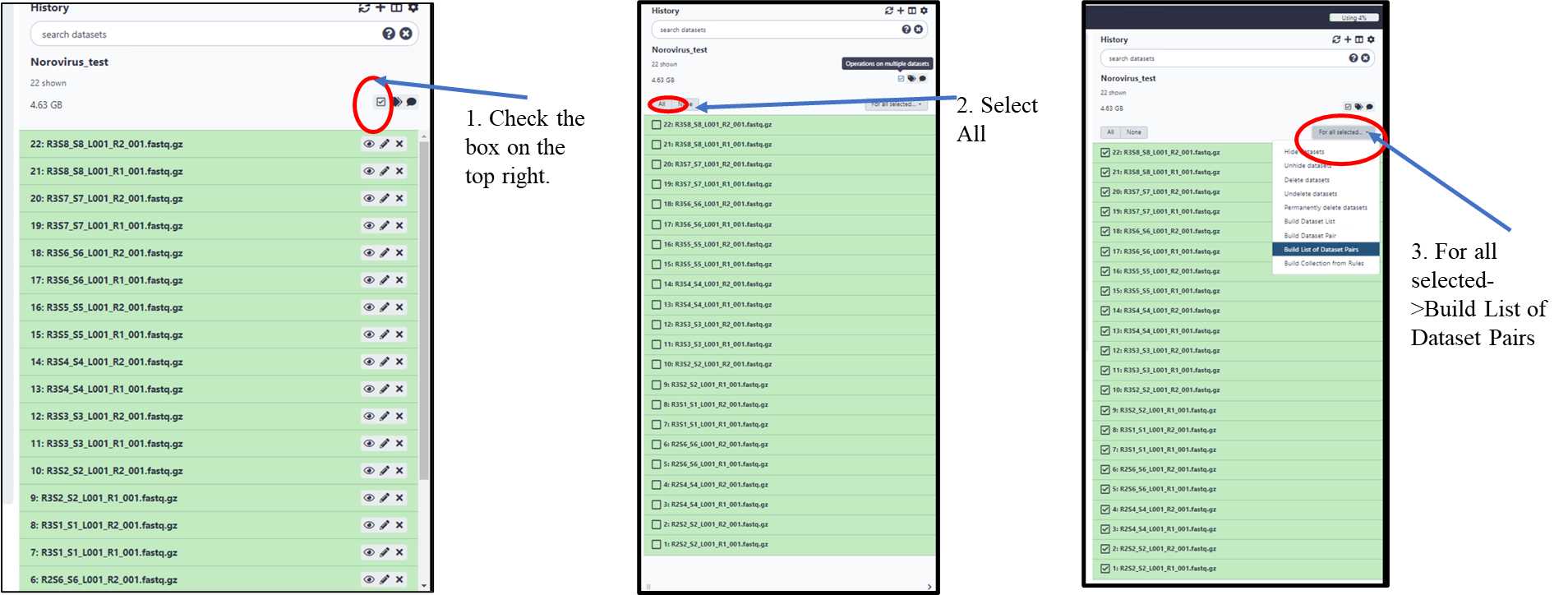
3.1.5. Build a list of Dataset Pairs (pairing the forward and reverse files into their respective samples for batch analysis (Follow steps1, 2 and 3).
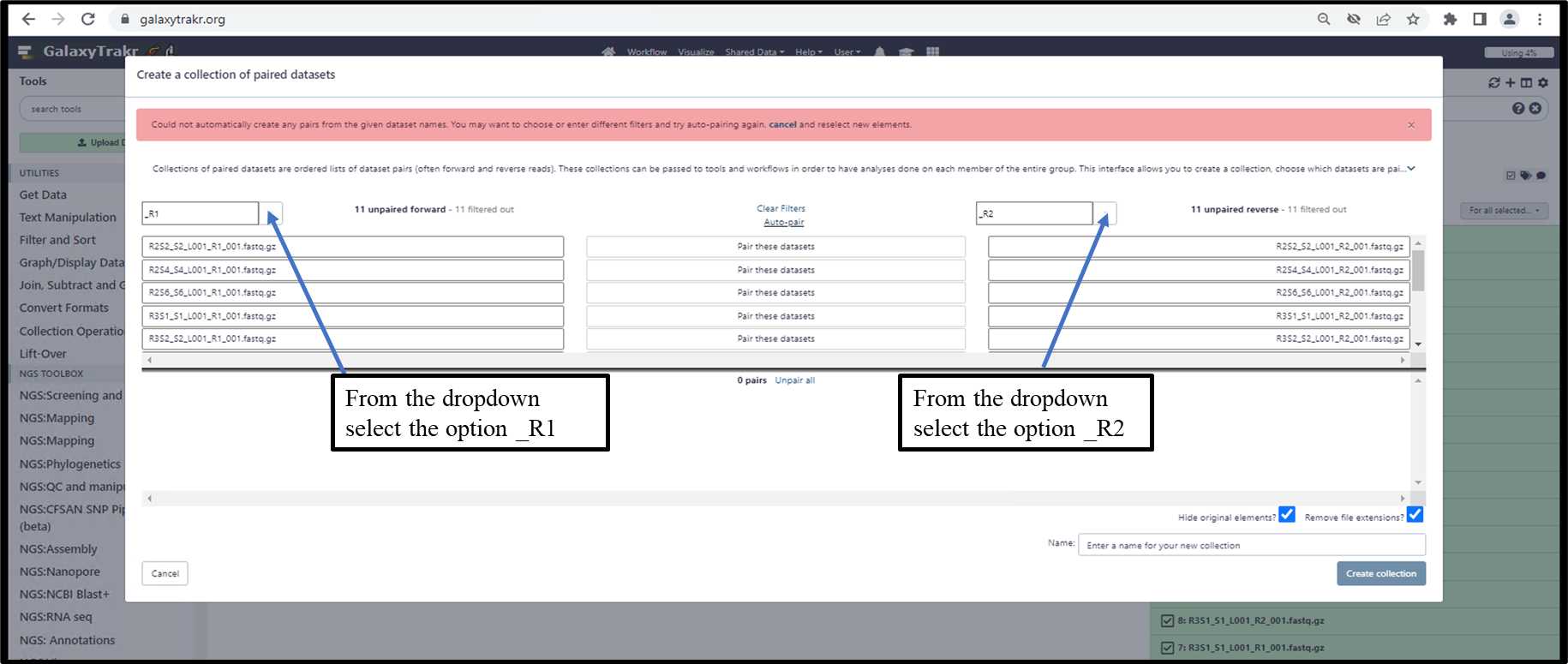
3.1.6. Create a collection of paired datasets.
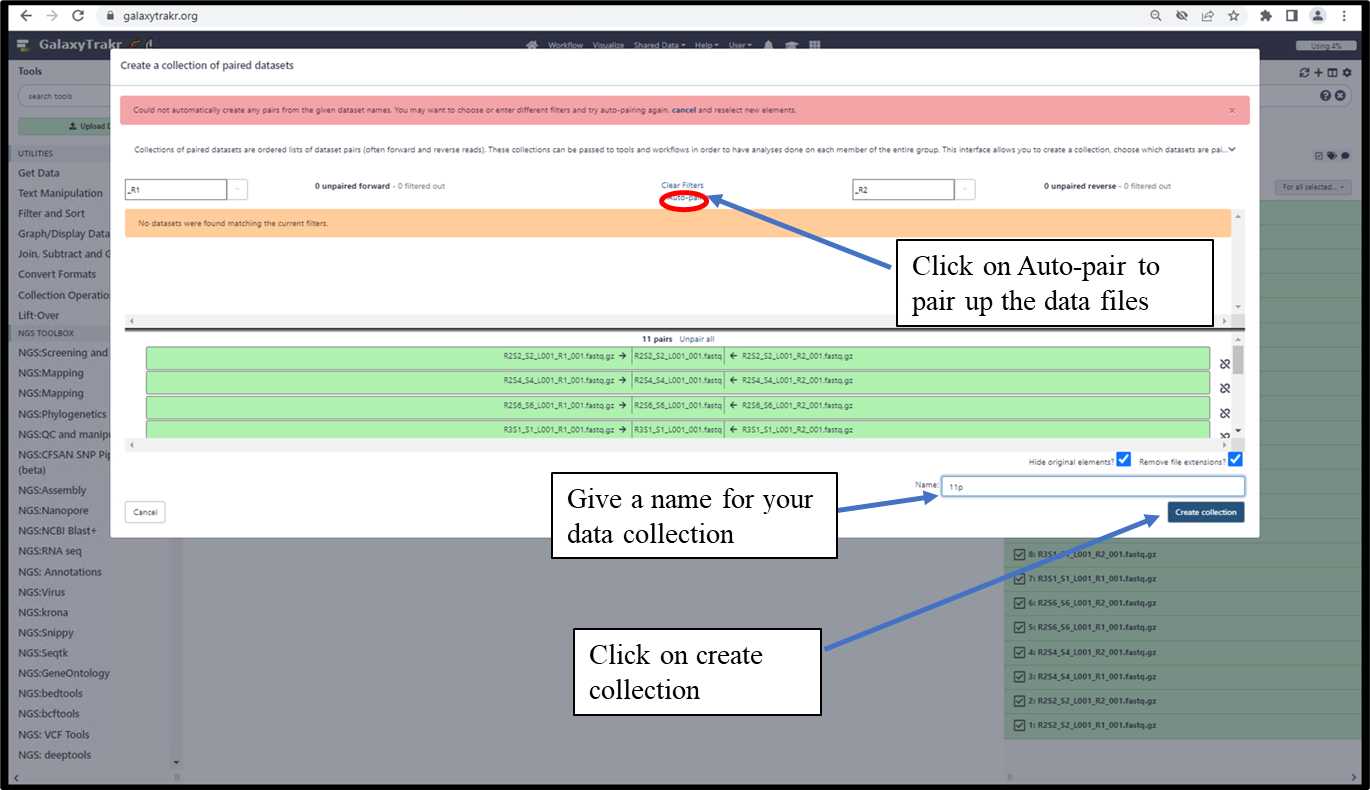
3.1.6. Create a collection of paired datasets - Cont.
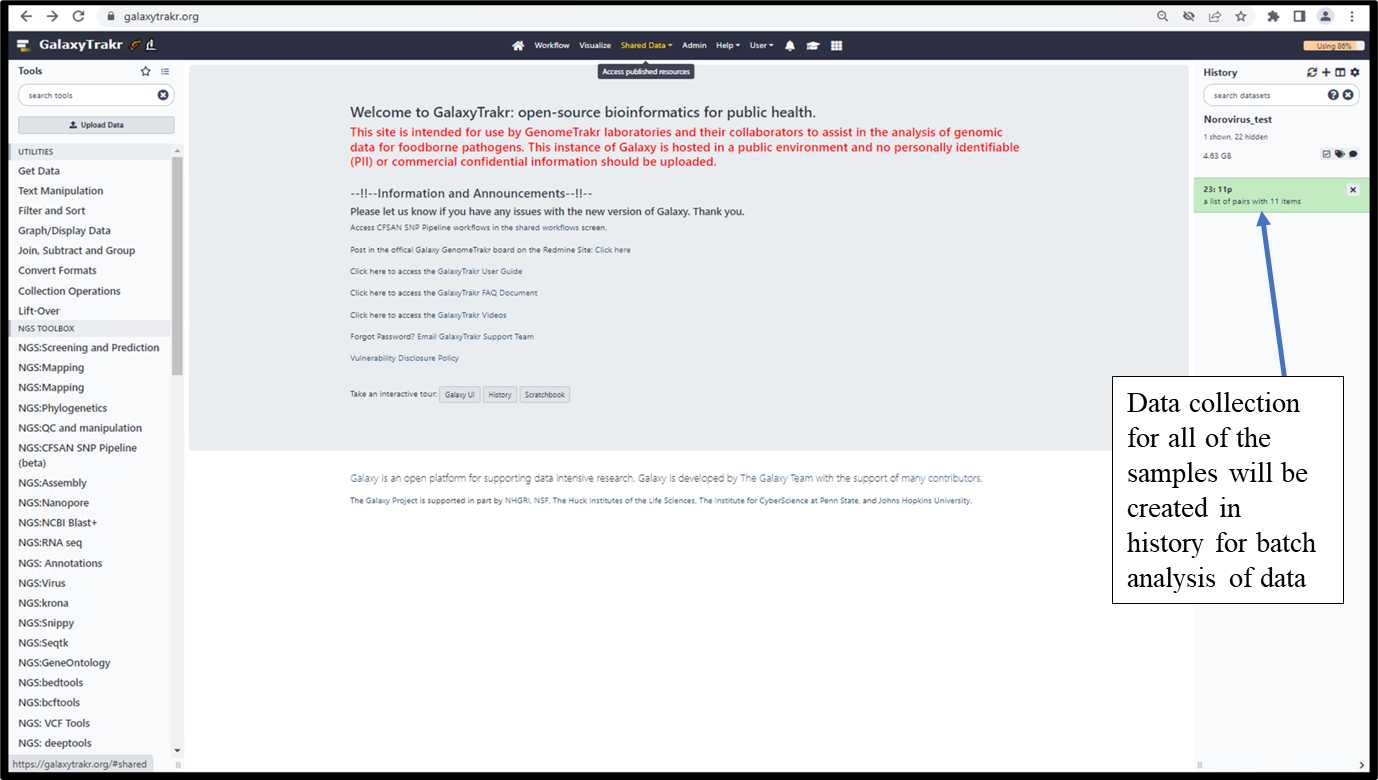
3.1.7. Data collection will be created in history.
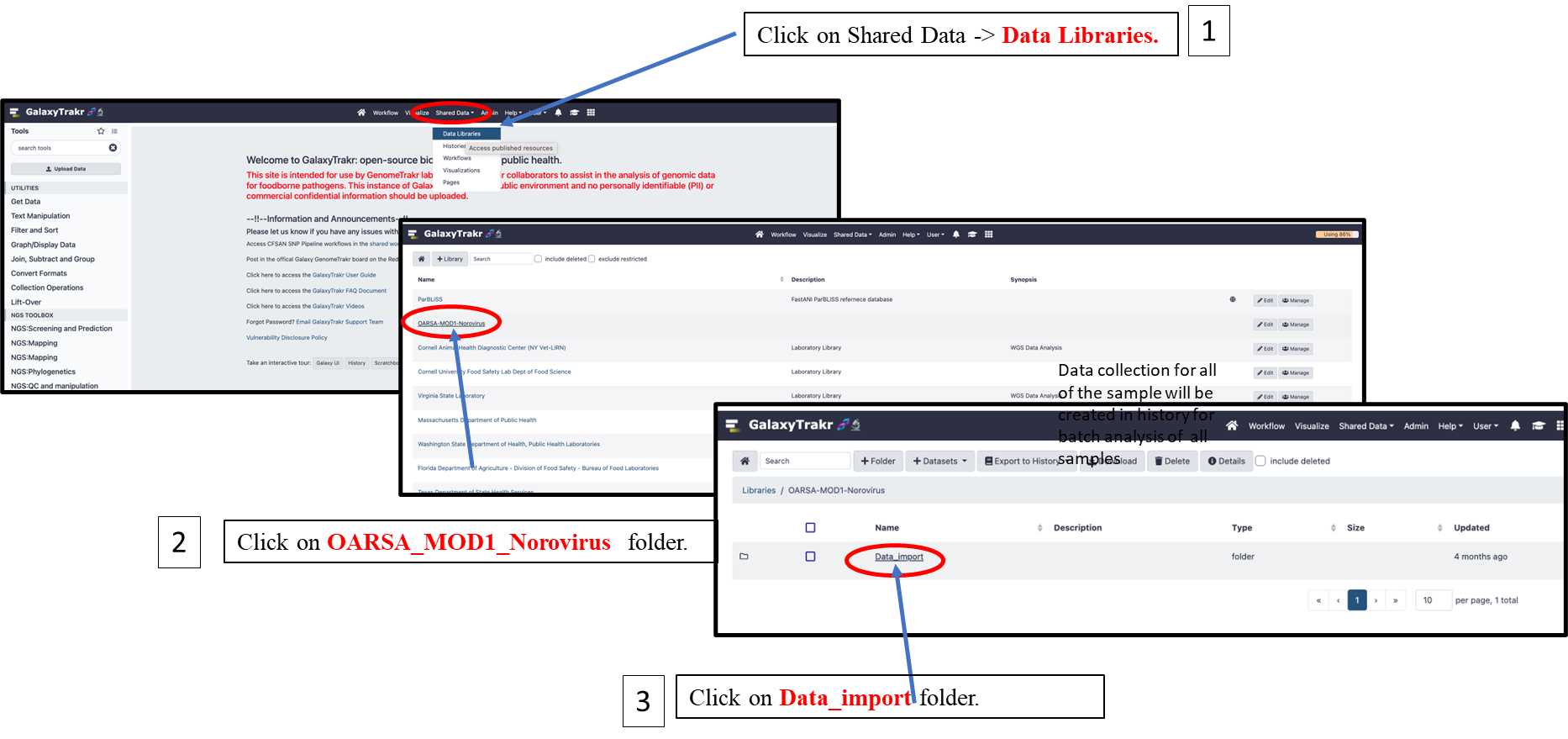
3.1.8. Import the reference data files from Shared Data folder following the steps 1-3 as shown below.
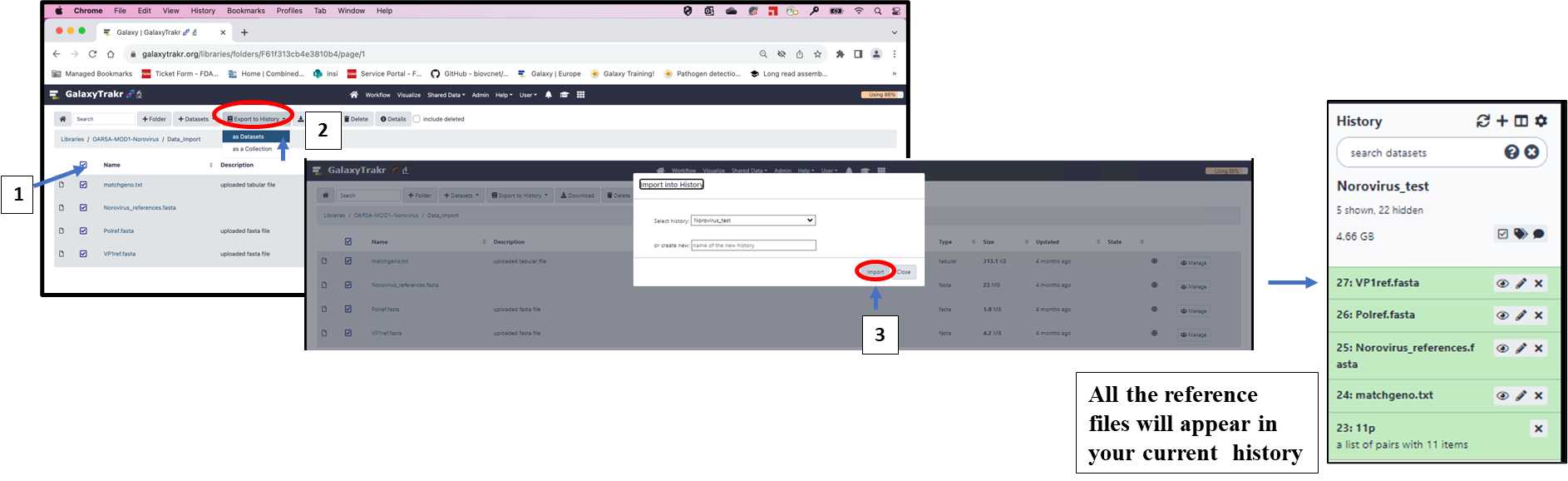
3.1.9. Select all files from the folder and export them as Datasets to your current history following the steps 1-3 as shown below.
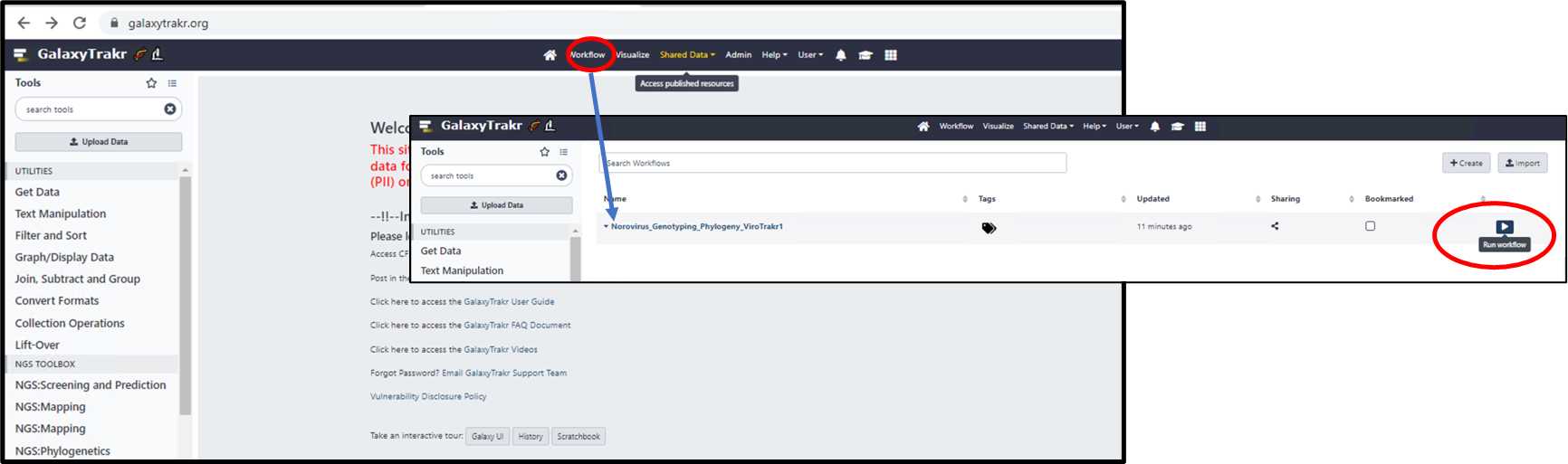
3.1.10. Click on WorkFlow tab from main menu, select and run the Norovirus_ Genotyping_Phylogeny
workflow.
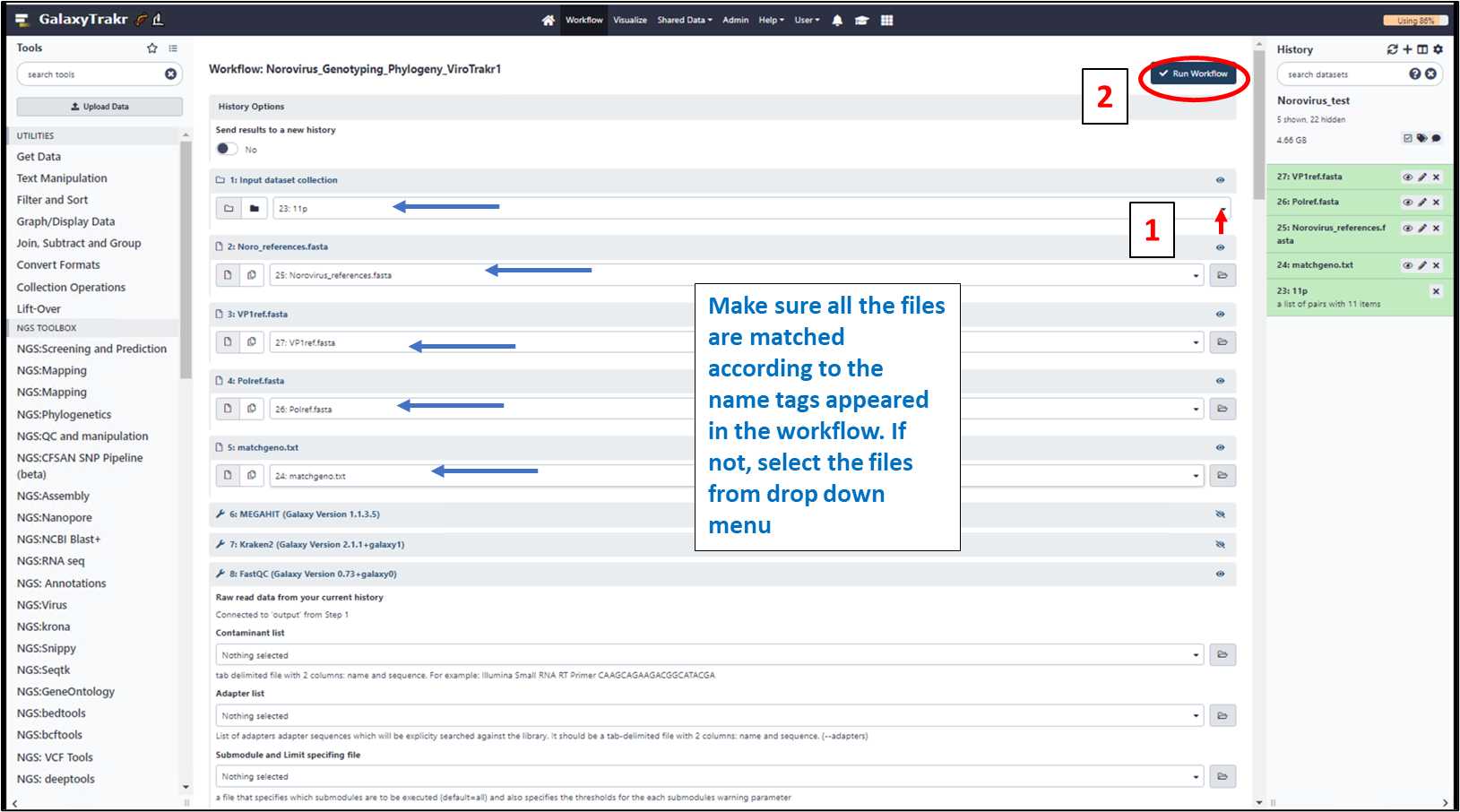
3.1.11. Select all the appropriate files from each dropdown menu and run workflow.
3.1.12. Once the workflow run is successful (Green status), results will appear in the
history.
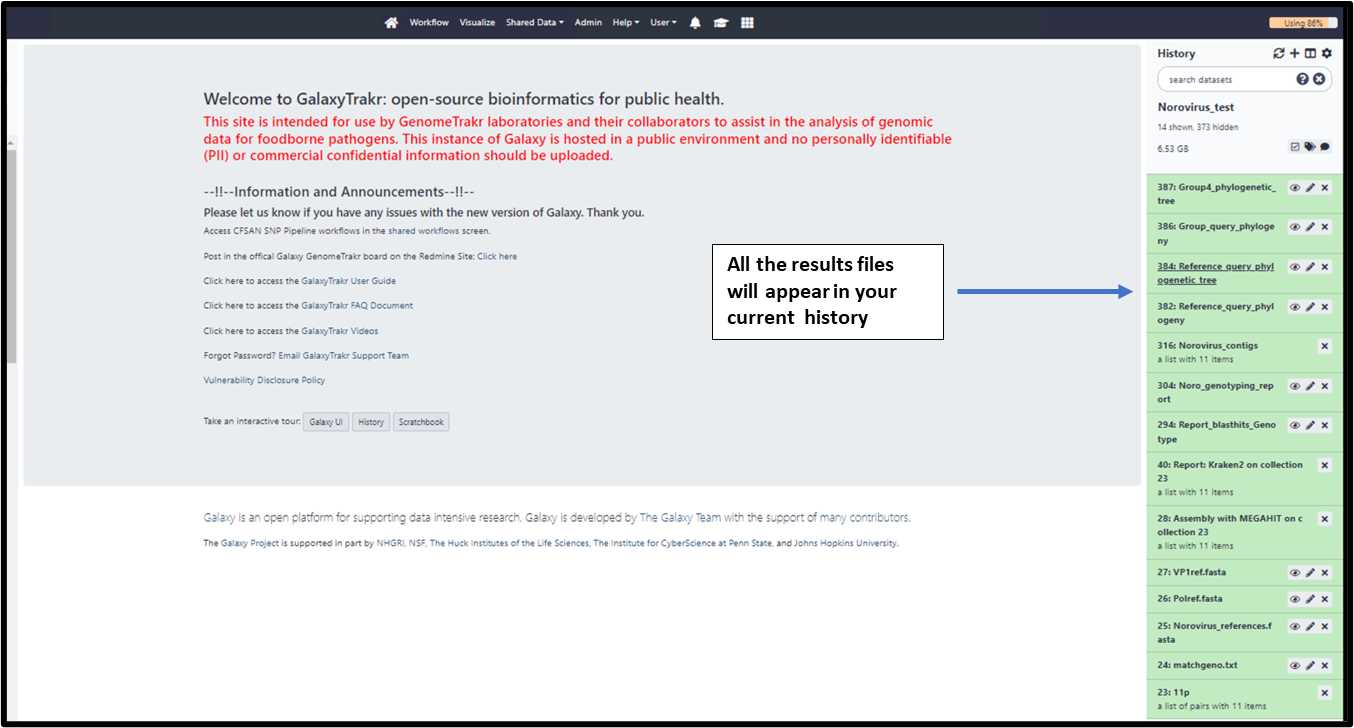
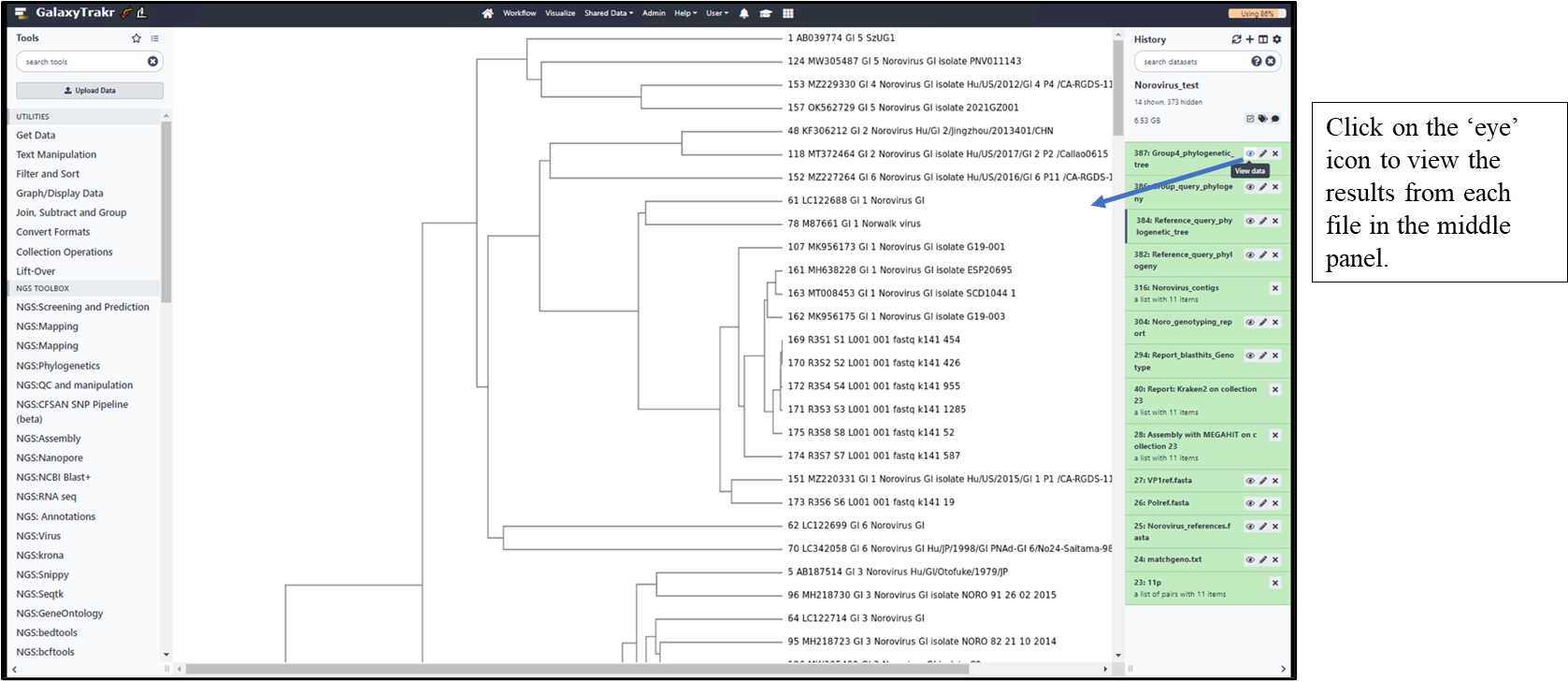
3.1.13. Select and view the result files in the middle panel.
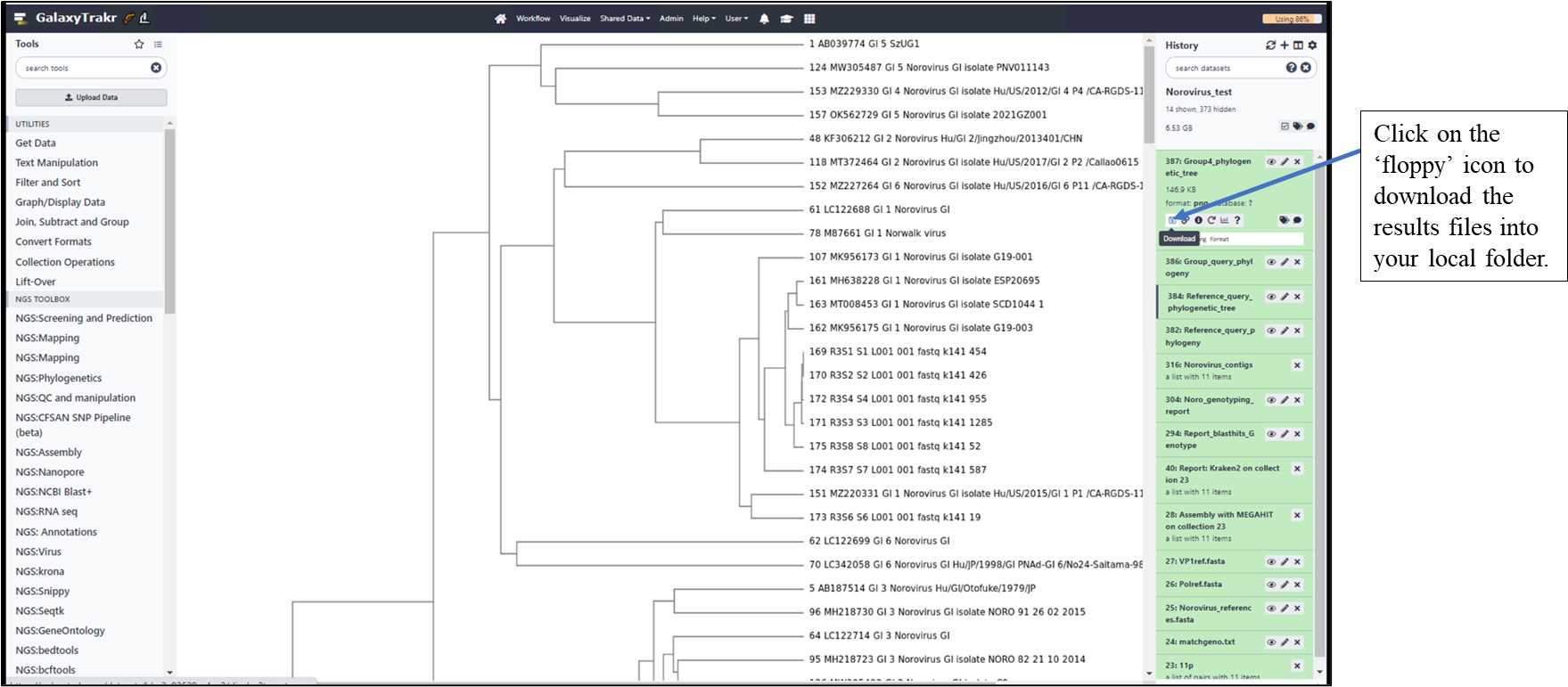
3.1.14. Download the result files to your local folder.
Result files include:
•Assembly with MEGAHIT: Metagenomic assemblies
•Report: Kraken2: Kraken2 reports
•Report_blasthits_Genotype: Reporting Best BLAST Hits against reference sequences
•Noro_genotyping_report: Final report contains QC stats and genotyping results
•Norovirus_contigs: Norovirus specific contigs extracted from metagenomics assembly
•Reference_query_phylogenetic_tree: Phylogenetic tree represents the input genomes along with the reference genomes from all groups .png format and .txt format.
•Group4_phylogenetic_tree:Phylogenetic tree represents the input genomes along with the GroupII.4 reference genomes from all groups .png format and .txt format.
Download data from SRA database (if you submit your raw data to ViroTrakr before GalaxyTrakr analysis).
3.2.1. SRA database link to norovirus sequence files for bioprojectPRJNA490509:
•https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP173043&o=acc_s%3Aa
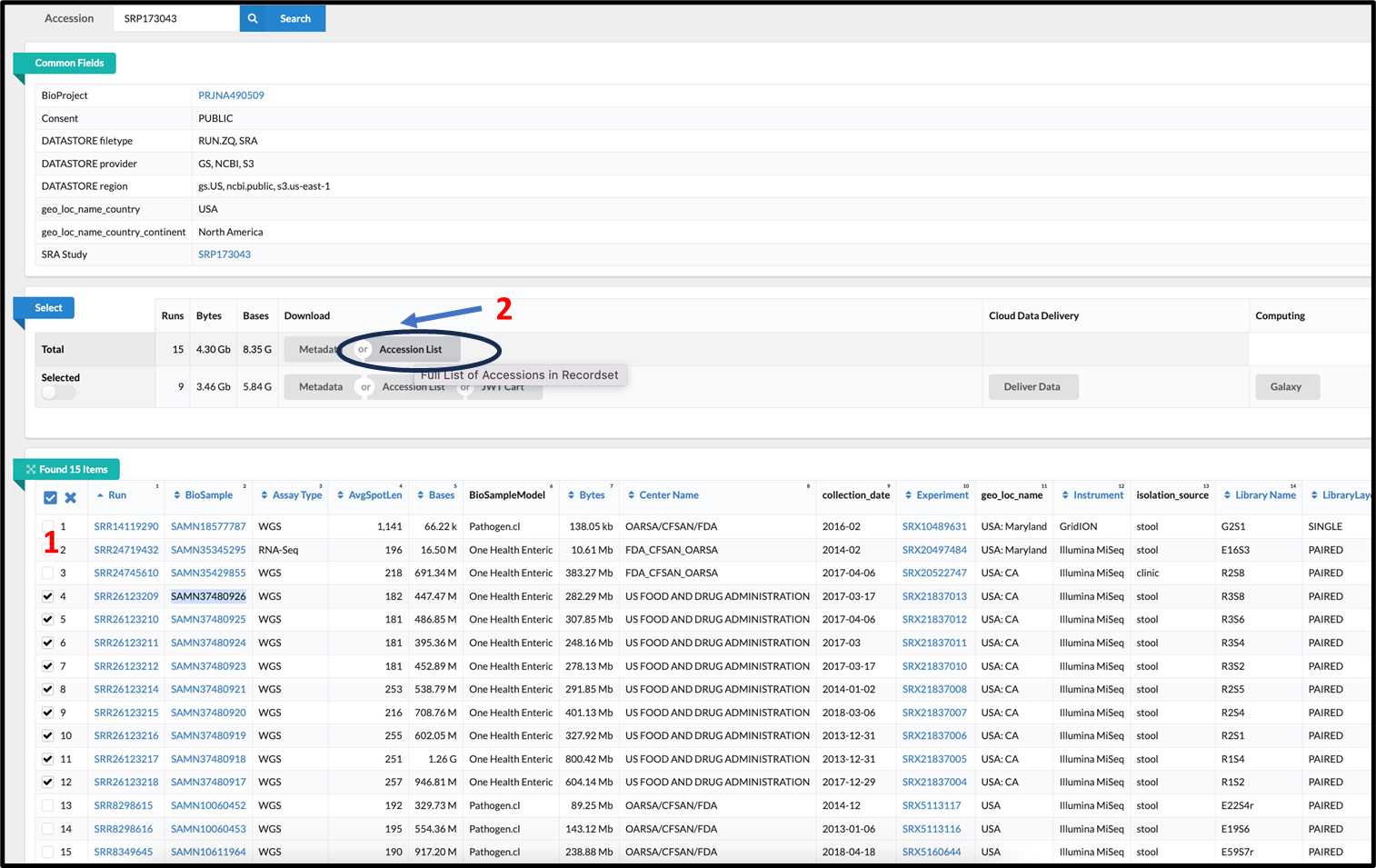
3.2.2. From SRA database, (1) select SRR samples of your choice then (2) click on accession list.
3.2.3.
The text files will be downloaded with SRR accessions in a format as SRR_Acc_List.txt:
For example: SRR26123209SRR26123210SRR26123211SRR26123212SRR26123214SRR26123215SRR26123216SRR26123217SRR261232183.2.4. To “Upload” the data, select the SRR_Acc_list.txt and click the “Start” button: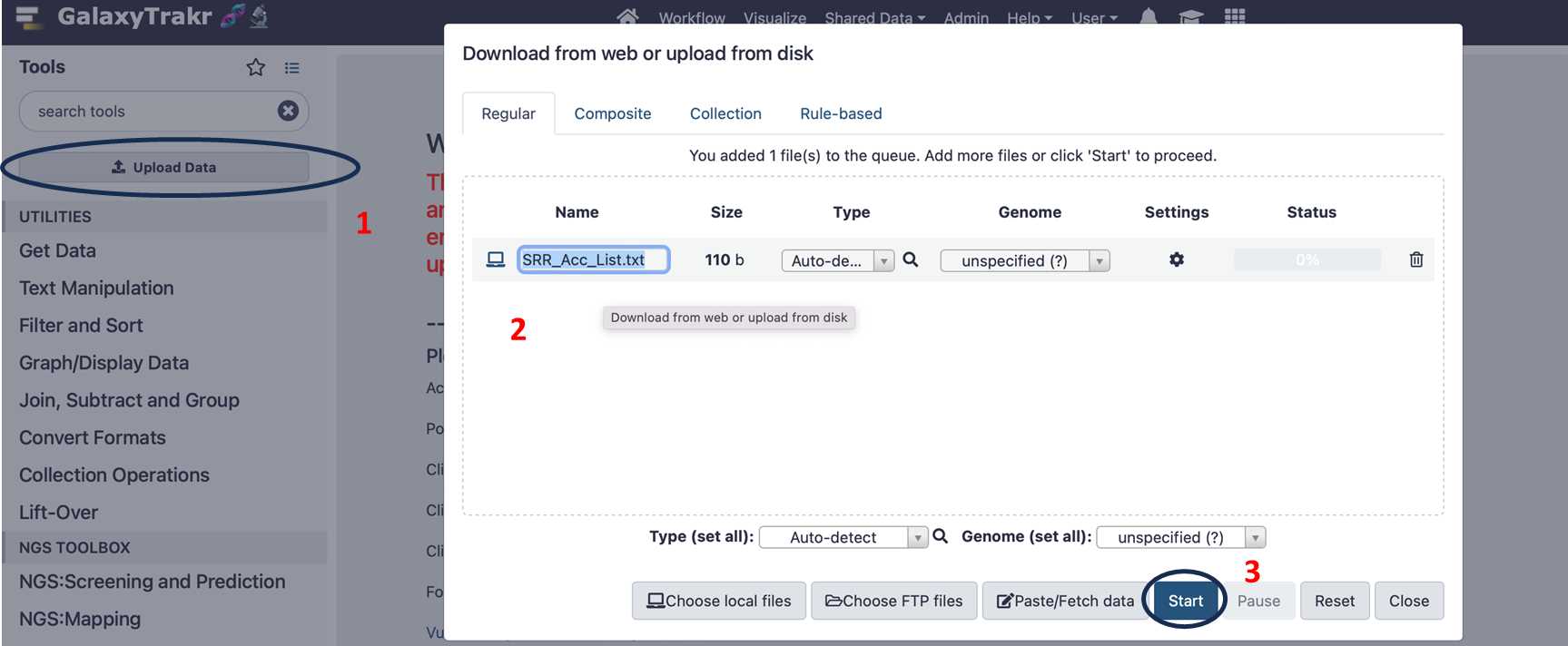
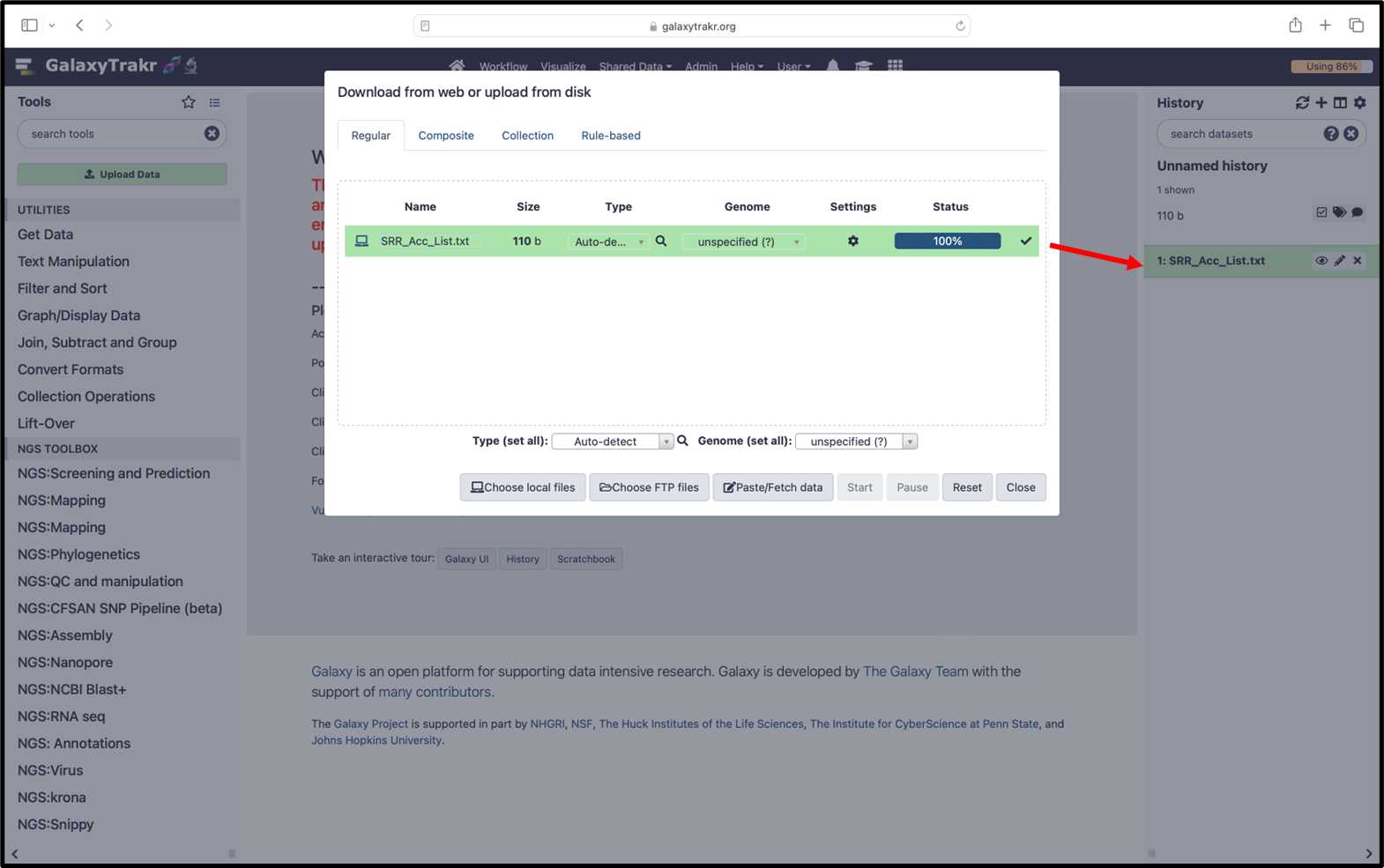
3.2.5. Once the download is completed, the text file will be added in history.
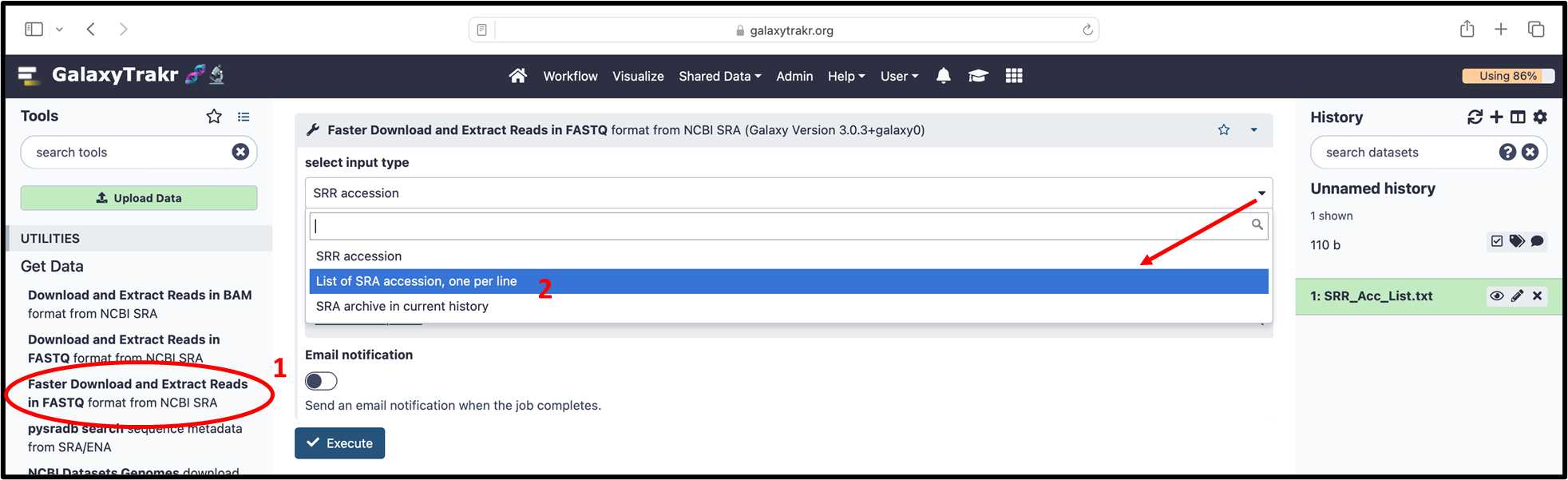
3.2.6. From “Get Data” on the left menu, select “Faster Download and Extract reads in FASTQ from NCBI SRA”, and select option by clicking on drop down menu “List of SRA accessions, one per line”.
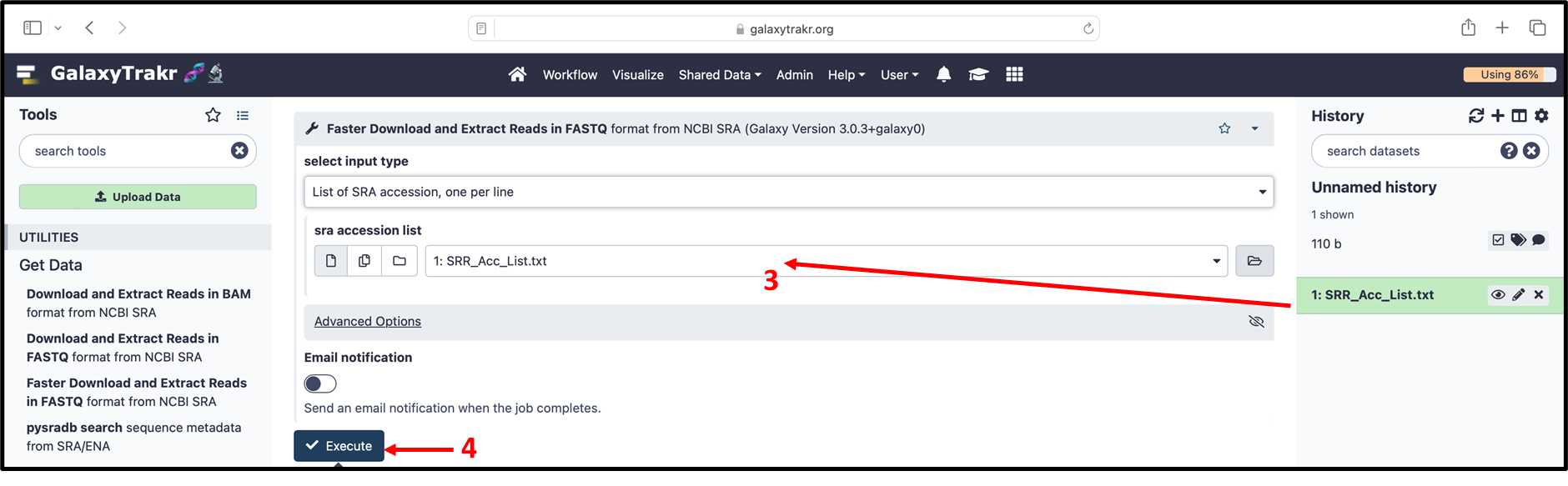
3.2.6. (cont.) Select the file SRR_Acc_List.txt and Click the Execute” button.
3.2.7. Data files will be downloading from NCBI SRA database. (Download time varies, depending on the number of files downloading and the NCBI server status).
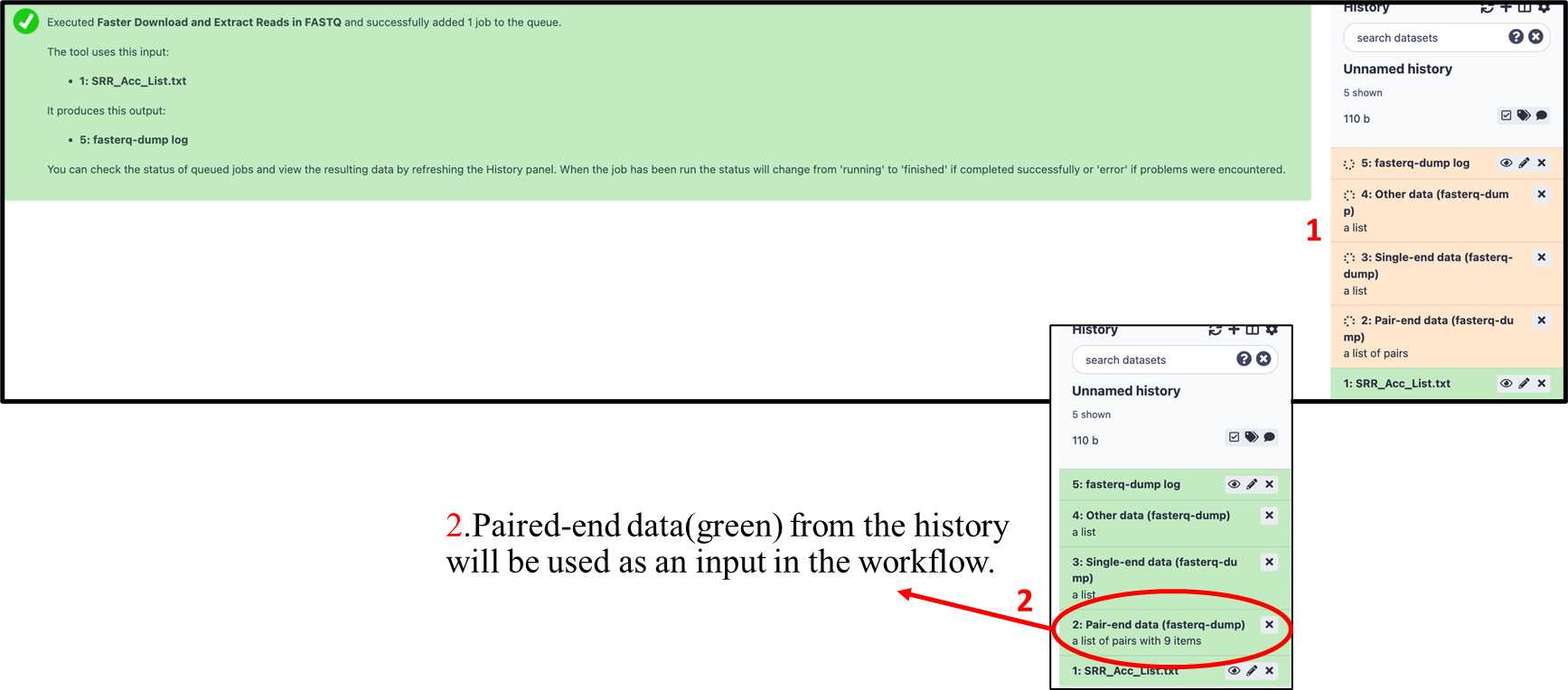
3.2.8. Follow the steps from 3.1.8. to 3.1.14 to run the workflow and collect the results.
