Library construction for human placenta bulk RNAseq
Scott Lindsay-Hewett
Abstract
This protocol describes the generation of stranded RNA-seq libraries from placenta total RNA. Since RNA quality can be an issue with total RNA isolated from placental tissue, it is advisable to enrich mRNA using a ribodepletion method rather than polyA selection.
Prior to ribodepletion, it is especially important to ensure that the input RNA is free of contaminating DNA. This protocol, therefore, begins with DNase treatment of samples, using Ambion's DNA- free DNase Treatment and Removal Reagents. Next, ribodepletion and library construction are performed using the KAPA RNA HyperPrep Kit with RiboErase (HMR). Libraries are indexed with KAPA Unique Dual-Indexed Adapters to enable multiplexed sequencing on an Illumina instrument.
Steps
DNA removal
To remove any possible contaminating DNA prior to ribodepletion, follow Ambion's protocol (Publication # 1906M, Revision E) for their DNA- free DNase Treatment and Removal Reagents.
DNA-free™ Kit DNase Treatment and Removal Reagents User Guide.pdf
Quantitate DNase-treated total RNA using Qubit RNA Broad Range Assay.
Ribodepletion and library construction
Follow KAPA's protocol (KR1351 - v4.21) for their KAPA RNA HyperPrep Kit with RiboErase (HMR). In our hands, this protocol yields great results with partially degraded total RNA samples from placenta, some with RIN scores as low as 2.
KAPA RNA HyperPrep Kit with RiboErase (HMR) KR1351 - v4.21.pdf
For HuBMAP samples, the following parameters were used:
Input total RNA : 1000ng
Fragmentation : tailored to RIN score (RIN score 6.0 and above, 0h 4m 0s``85°C; RIN score 5.0-5.9, 0h 3m 30s``85°C; RIN score 4.9 and below, 0h 3m 0s``85°C)
Library amplification : 8 cycles
For indexing, and to enable efficient multiplexed sequencing, use the KAPA Unique Dual-Indexed Adapter Kit , and dilute adapters to 7micromolar (µM). Using UDIs reduces "index hopping" which can be an issue when using combinatorial dual indexes, especially on patterned flow cells like the NovaSeq.
KAPA Unique Dual-Indexed Adapter Kit KR1736 - v3.20.pdf
Quality control
Quantitate libraries using the Qubit DNA High Sensitivity Assay, and check library distribution by running the DNA High Sensitivity Assay on an Agilent Bioanalyzer. A typical trace is shown below.
. These primer dimers are not an issue since they cannot cluster on the flow cell.")
If multiplexing samples, first perform a balancing run to ensure equal representation of all samples in the pool. For the balancing run, prepare an "equal volume" pool by combining 2µL each library together. Run the pool on a MiSeq instrument using a MiSeq Reagent Kit Nano. Based on the proportion of reads assigned to each index during the Nano run, prepare a balanced pool that will yield an equal read depth for all samples in the pool.
Prior to submitting for sequencing, quantitate the pool using the Qubit High Sensitivity DNA assay. Determine the average fragment size for each library from the Bioanalyzer traces.
Determine the average fragment size in the balanced pool, and use the following formula to determine the nM concentration:
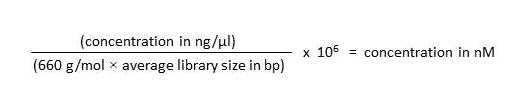
Submit the pool to your sequencing facility, noting the nM concentration.
For HuBMAP bulk RNA-seq samples, the multiplexed pool was sequenced on a NovaSeq 6000 S4 lane using a 100bp paired-end run configuration.
