"Midnight" SARS-CoV2 genome sequencing protocol using 1200bp amplicon primer set v2 and the Nanopore Rapid library kit
Benjamin Schwessinger, Nikki Freed, Lydia Murphy, Olin Silander
Abstract
This protocols is part of the ANU Biosecurity mini-research project #2 "An SARS-COV2 incursion scenario: Genomics, phylogenetics, and incursions." This mini-research project is modeled on the yearly Quality Assurance Program of The Royal College of Pathologists of Australia (RCPAQAP), we take part in together with ACT Pathology.
This research project is split into two major parts, identical to how the official RCPAQAP is run every year.
Part #1 is focusing on the 'wet- lab' by sequencing SARS-COV2 from real world RNA samples provided by ACT Pathology especially for our ANU biosecurity course (Thank YOU!). Here you will amplify and sequence five (5) RNA samples per research group. You will assess the SARS-COV2 genome sequences for their lineage assignments using online programs, put sequences into a global context, estimate the collection date based on genetic information, and describe mutations in the spike protein.
Part #2 is focusing on the 'dry-lab' by investigating a hypothetical incursion scenario in the so-called city Fantastica. You will combine genomic surveillance of SARS-COV2 with case interview data to trace the spread into of SARS-COV2 in the community and into high risk settings. We will provide you with real publicly available SARS-COV2 genome and fantasized case interviews. You will put these two together to trace the spread and suggest potential improvements in containment strategies with a focus on high risk settings.
This protocol describes the wet-lab component of Part #1. It is an adaption (fork) of a previous protocol published by Nikki Freed and Olin Silander during the early days of the pandemic here and here. They designed the protocol to enable faster, easier sequencing of SARS-COV2 genomes with fewer steps than previous methods, we use multiplexed 1200 base pair PCR amplicons with the Oxford Nanopore RAPID barcoding kit.
The original publication that describes this protocol is here along with important considerations is here: https://academic.oup.com/biomethods/article/5/1/bpaa014/5873518?login=true
Primers were all designed using Primal Scheme: http://primal.zibraproject.org/, described here https://www.nature.com/articles/nprot.2017.066.
You will be using the midnight primer set v2 described here and purchased here https://sg.idtdna.com/pages/products/next-generation-sequencing/workflow/xgen-ngs-amplicon-sequencing/predesigned-amplicon-panels/sars-cov-2-midnight-amp-panel.
The final goal is to achieve the following:
Week 7:
- cDNA reverse transcription of RNA into DNA.
- Pooled amplification of the SARS-COV2 genomes chunked into ~1200bp amplicons with non-overlapping pools A and B.
Week 8:
- Library preparation using the Nanopore Rapid barcoding kit.
- Sequencing your own library on a Nanopore flongel.
This protocol is applicable for week 7 and 8.
Before start
You must study the protocol carefully before you start. If anything is unclear post questions directly here on protocols.io.
Steps
Week 7: cDNA synthesis and two pooled PCR reactions
In week 7 you will perform the cDNA synthesis and PCR reactions on five samples and one negative control. You will be provided with five samples of 10 ul each and one tube of negative control.
cDNA preparation
You will receive a PCR strip of eight with 6 tubes containing 2uL Lunascript RT supermix. Label the tubes with 1-6 and your RG name. Note down which sample corresponds to which number.
Mix the following components in an 0.2mL 8-strip tube on ice;
Component Volume
LunaScript®RT SuperMix Kit (5x) 2µL
Template RNA 8µL
**Total** `10µL`
This means add 8uL of sample to each tube separately. Gently mix by pipetting up and down during the adding of each sample. Change the tip between each sample addition. Once you added all samples close the lid.
Pulse spin the tube to collect liquid at the bottom of the tube.
Incubate the reaction in a PCR machine:
25°C 0h 2m 0s
55°C 0h 10m 0s
95°C 0h 1m 0s
Hold at 20°C
PCR amplification with two primer pools
You will have to prepare two PCR master mixes one for each primer pool A and B. For this, you will have two tubes of 81.25ul Q5 hot start master mix provided. Label each tube with pool A or B and your RG name.
Component and volume per sample Pool A Pool B
Q5® Hot Start HF 2x Master Mix 12.5µL 12.5µL
Primer Pool A (10µM) 1.1µL 0µL
Primer Pool B (10µM) 0µL 1.1µL
Nuclease-free water 8.9µL 9.0µL
**Total** `22.5µL` `22.5µL`
Considering you have five samples and one negative control, generate the following two master mixes.
| A | B | C |
|---|---|---|
| Q5® Hot Start HF 2x Master Mix (two tubes provided) | 81.25ul | 81.25ul |
| Primer Pool A (10µM) | 7.15ul | 0 |
| Primer Pool B (10µM) | 0 | 7.15ul |
| Nuclease-free water | 57.85ul | 57.85ul |
Once you added all ingredients, mix by vortexing and spin down to collect the liquid. Place on ice.
Label two PCR strips of eight with the PCR pool A or B, number 1-6, and your RG name. Aliquot out 22.5 ul of each master mix (Pool A or B) into the corresponding PCR strips.
Collect your cDNA containing strip tubes (the samples will have a blue color). Spin them down briefly.
add 2.5µL each cDNA sample [1-6] to a tube containing 22.5µL Pool A that is labelled accordingly . Mix well by pipetting up and down. Change tip after each addition.
Add 2.5µL each cDNA sample [1-6] to a tube containing 22.5µL Pool B that is labelled accordingly . Mix well by pipetting up and down. Change tip after each addition.
Leave the strip tubes on ice. Close the tubes with a lid when done adding all samples.
Pulse centrifuge the tubes to collect the contents at the bottom of the tube.
Set-up the following program on the thermal cycler:
Step Temperature Time Cycles
Heat Activation 98°C 0h 0m 30s 1
Denaturation 98°C 0h 0m 15s 35
Annealing and Extension 65°C 0h 5m 0s 35
Hold 20°C Indefinite 1
Hand over your PCR reactions to demonstrators as they will run them till next week.
Week 8: Library preparation and sequencing
In week 8 you will perform your own Nanopore sequencing library preparation and sequencing. You will do this by using your PCR reactions from week 7.
Pooling and PCR quantification
Label a 1.5mL Eppendorf tube with sample name and RG name. In total this will be six tubes.
For each sample combine the PCR reactions (pool A and B) by pipetting all 25ul of each pool into one tube. This means each 1.5 ml eppendorf tube will contain 50ul in total.
Mix by vortexing and spin down samples briefly.
Quantify DNA using a Qubit.
You will be provided with 6 tubes of 198ul Qubit HS solution.
Label the tubes with the numbers 1-6.
Add 2uL of each combined PCR reactions into one Qubit tube.
Vortex briefly, hand centrifuge, incubate for 2 mins, and measure with the Qubit machine.
Note down your concentration in ng/ul for each sample.
Normalisation
Label a 1.5 eppendorf tube for each sample so you can generate a stock PCR solution of 13.33 ng/ul the final volume being 20uL.
Each tube must be labelled as stock 13.33 ng/ul, sample name, and RG name.
The important formula here is:
n = c*V with n being the amount in ng, c the concentration in ng/ul, and V the volume.
You want V = 20ul and c =13.33 ng/ul. This means you will have 267ng in 20 ul.
The question is how much volume of the original PCR do you need to obtain 267ngs?
Here now n = 267g and c equals your measured concentration. Let's assume your measured c = 56 ng/ul for an assumed sample. Than we can resolve the equation as follows.
n = c*V -> V = n/c -> 267ng/56ng/ul = 4.7 ul
So you need to use 4.7ul in 20ul total volume to obtain 267ng. This means you can combine 13.7ul H20 with 4.7ul of your combined PCR sample. Vortex it and spin down.
In case you concentration is < 13.33 ng/ul use 20ul of sample as stock without dilution.
Rapid barocoding using the SQK RBK110.96
Mulitple samples can be run on the same flow cell by barcoding. Up to 96 samples at a time can be sequenced using the RBK110.96 kit. Amplicons from each sample will be individually barcoded in the following steps.
You will receive a PCR strip of eight with six tubes containing 2.5ul of the fragmentation mix each. Label the strip with sample number [1-6] and RG name.
Add 7.5µL of each diluted PCR stock [13.33 ng/ul] reaction from step 12 to the labeled PCR tube.
Set up the following reaction for each sample:
Component Volume
DNA amplicons from step 12 (100ng total) 7.5µL
Fragmentation Mix (one for each sample, already aliquoted for you) 2.5µL
Total 10µL
Important keep samples on ice!
Mix gently by pipetting up and down briefly, flick the tubes, and spin down.
Incubate the reaction in a PCR machine:
30°C for 0h 1m 0s
80°C for 0h 1m 0s
4°C for 0h 0m 30s
From here on out you will combine your barcoded samples with one other RG on your bench. Each final library will contain 12 samples to be sequenced together. [With the exception of RG0 who have a single flongel to themselves]. RG1+2, RG3+4 and so on will go together.
For each pair of RGs, label one 1.5 ml Eppendorf tube with BC and RG names. Combine all your barcoded samples in this tube. This tube will contain 120ul total. [RG0 will have 60uL]
Mix by vortexing and spin down.
SPRI Bead Cleanup. Use a 1:1 ratio of sample to beads. This means in case of 120ul combined barcoded samples add 120ul of beads. Ask for help of a demonstrator as needed.
Add 120uL of SPRI beads to your combined barcoded samples. Mix by flicking the tube. Hand centrifuge to collect all liquid in the bottom of the tube while the brown beads stays suspended.
Remove the tube from the magnetic rack. Resuspend pellet in 15µL Elution Buffer (EB). Incubate for 0h 3m 0s at RT. with flicking the tube.
Label a new 1.5ml tube with BCD and your RG names.
Place on magnet, incubate for 0h 3m 0s so that all beads are on the side of the tube towards the magnet. Transfer 10µL to a clean labelled 1.5mL Eppendorf tube ensuring no beads are transferred into this tube.
Incubate for 0h 5m 0s at room temperature.
Place tube on magnetic rack and incubate for 0h 3m 0s until the beads have pelleted towards the magnet and the supernatant is completely clear.
Leave tube on the magnet and pipette off the supernatant, being careful not to touch the bead pellet. The DNA is now bound to the beads.
Add 1ml of freshly prepared room-temperature 80% volume ethanol to wash the pellet.
and repeat one more ethanol wash.
Leave tube on the magnet and pipette off the supernatant, being careful not to touch the bead pellet. The DNA is now bound to the beads.
Pulse centrifuge to collect all liquid at the bottom of the tube, place tube back to the magnet, and carefully remove as much residual ethanol as possible using a P10 pipette.
With the tube lid open incubate for 0h 3m 0s to let the pellet try.
Label a new 1.5ml Eppendorf tube with L and your RG name.
Transfer 5µL of your cleaned combined barcoded samples into this tube.
Ask a demonstrator to add 0.5µL of Rapid Adapter F (RAP-F) to 5 μl of barcoded DNA.
Mix gently by flicking the tube, and spin down.
Incubate the reaction for 0h 5m 0s at RT.
Now prepare the final library to be loaded by combining the following a 1.5ml tube labelled FL and your RG name.
15µL Sequencing Buffer II (SBII)
10µLLoading Beads II (LBII) mixed immediately before use
2.5µL EB buffer
2.5µLof RAP-F adapted barcoded DNA from step 15.
Place library to the side till you are ready to load in step 23.
Loading the library onto a Flongel
Please familiarize yourself with how to load a flongel in this video https://nanoporetech.com/resource-centre/video/ncm22/how-to-load-a-flongle-flow-cell [0-12 minutes only]. Ask a demonstrator to help with the loading!
Place the Flongle adapter into the MinION.

The adapter should sit evenly and flat on the MinION. This ensures the flow cell assembly is flat during the next stage.
Plug in the MinION contain the flongle adapter in the computer.


Place the flow cell into the Flongle adapter, and press the flow cell down until you hear a click.
The flow cell should sit evenly and flat inside the adapter, to avoid any bubbles forming inside the fluidic compartments.
Take the tube labelled with FL that contains 117 μl of Flush Buffer (FB) with 3 μl of Flush Tether (FLT). Mix it by flicking an collect it at the bottom of the tube.
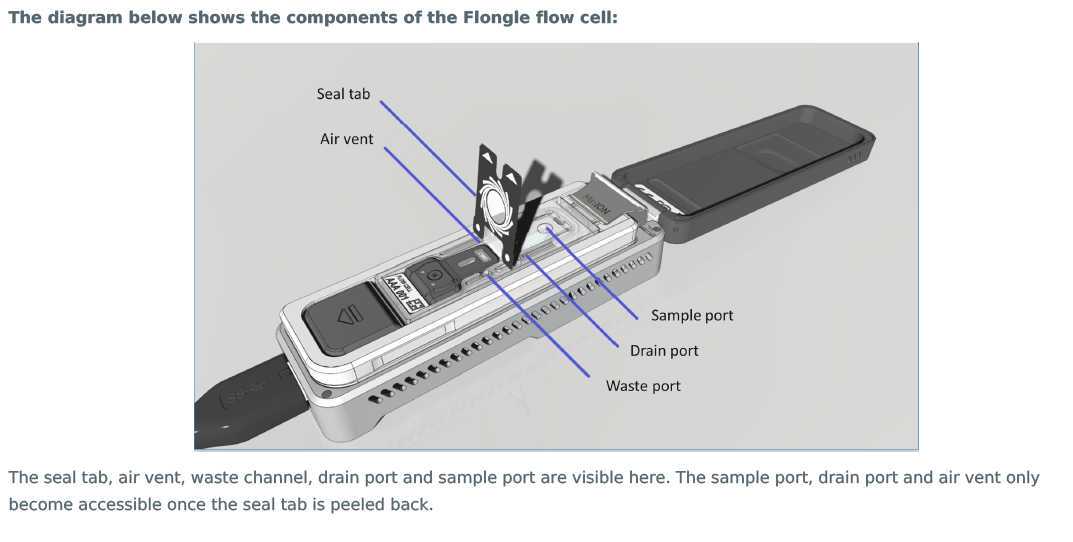
Peel back the seal tab from the Flongle flow cell, up to a point where the sample port is exposed, as follows:
- Lift up the seal tab:
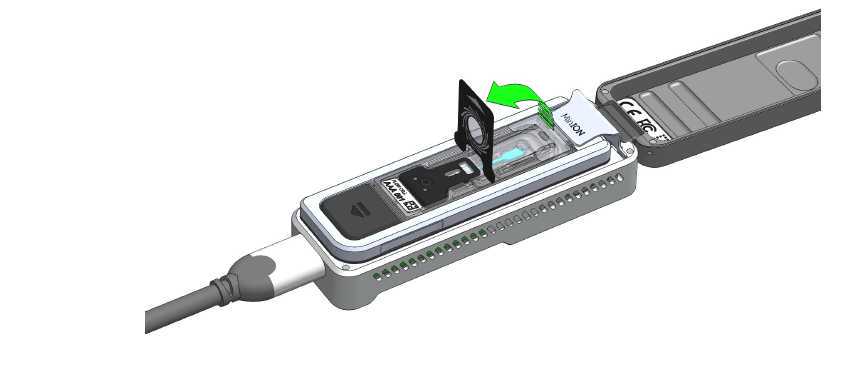
- Pull the seal tab to open access to the sample port:

- Hold the seal tab open by using adhesive on the tab to stick to the MinION Mk 1B lid:

Take the 120ul of FL. To prime your flow cell with the mix of Flush Buffer (FB) and Flush Tether (FLT) that was prepared earlier, ensure that there is no air gap in the sample port or the pipette tip. Place the P200 pipette tip inside the sample port and slowly dispense the priming fluid into the Flongle flow cell by slowly pipetting down. We also recommend twisting the pipette plunger down to avoid flushing the flow cell too vigorously.
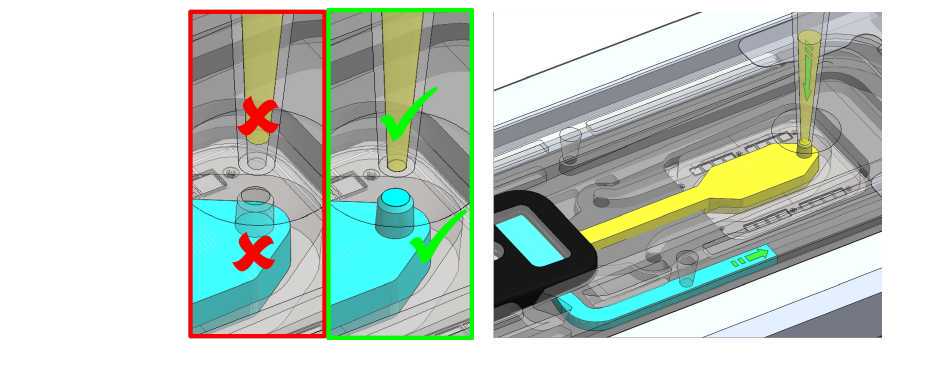
Take the final library from step 16 and mix by flicking so it is milky again. Collect by a quick hand centrifuge. Take the 30 ul and load.
To add the final library to the flow cell, ensure that there is no air gap in the sample port or the pipette tip. Place the P200 tip inside the sample port and slowly dispense the Sequencing Mix into the flow cell by slowly pipetting down. We also recommend twisting the pipette plunger down to avoid flushing the flow cell too vigorously.
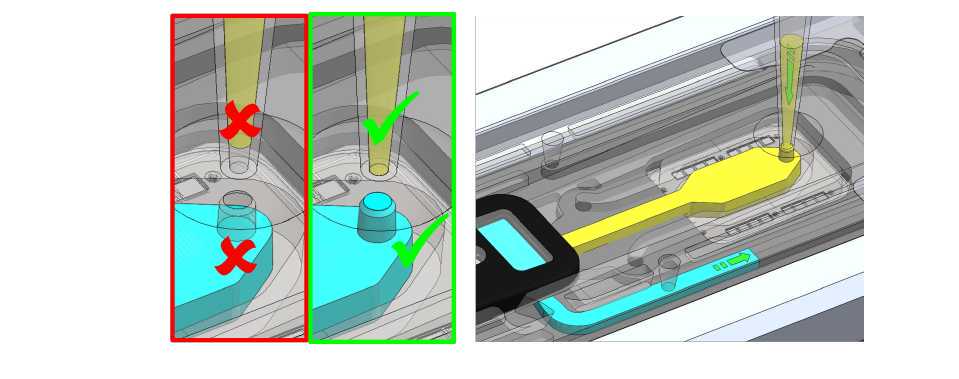
Seal the Flongle flow cell using the adhesive on the seal tab, as follows:
- Stick the transparent adhesive tape to the sample port.
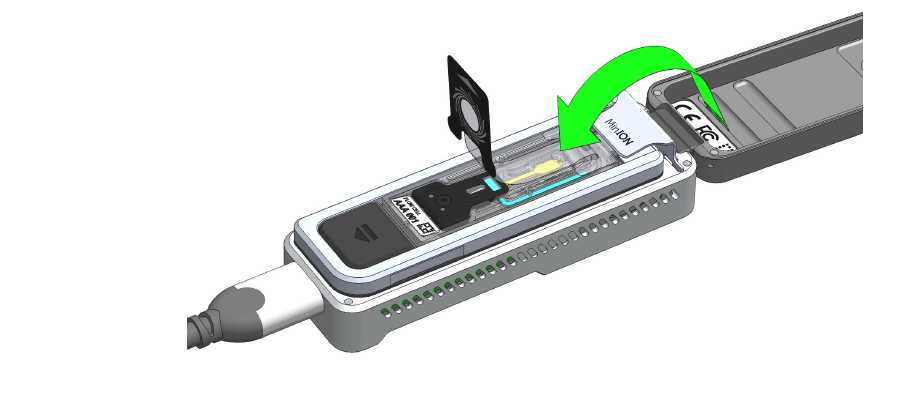
- Replace the top (Wheel icon section) of the seal tab to its original position.
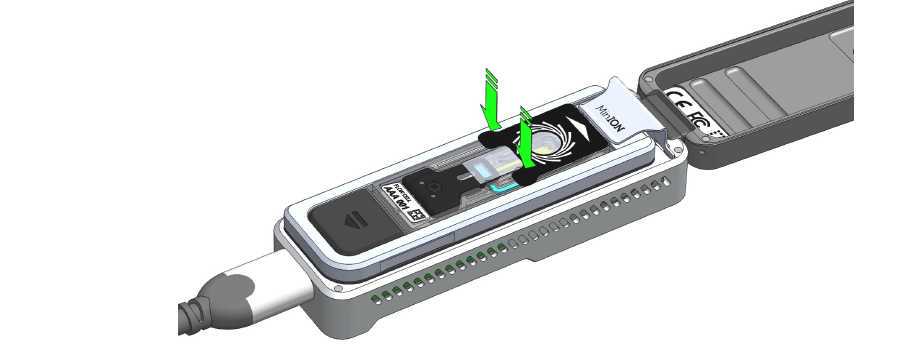
Close the MinION lid.
Data acquisition and basecalling
Open the MinKNOW software on the laptop and get a demonstrator to help you get the sequencing run started.
