Direct Detection of poliovirus and Nanopore Sequencing (DDNS) - Stool
Joyce Akello, Alex Shaw, Manasi Majumdar, Catherine Troman, Javier Martin, Nick Grassly, Aine OToole, c.ansley, Erika Bujaki, rachel.colquhoun, khurshida, alammu, Andrew Rambaut, arshady, Shannon Fitz, Ben Bellekom
Abstract
This protocol is an update from the protocol described in the paper "Rapid and sensitive direct detection and identification of poliovirus from stool and environmental surveillance samples using nanopore sequencing" by Shaw et al in the Journal of Clinical Microbiology (2020), DOI: 10.1128/JCM.00920-20 and is commonly known as Direct Detection of Poliovirus by Nanopore Sequencing (DDNS).
The protocol aims to amplify the VP1 region of poliovirus through a semi-nested PCR using a pan-Enterovirus primer and polio specific primers followed by amplification of the VP1 region using a polio specific primer set. We use barcoded primers as this greatly simplifies the subsequent library preparation process.
This protocol is for use with Oxford Nanopore kit14 chemistry ligation sequencing reagents and can be used with the MinION Mk1B or GridION sequencer.
Within the protocol steps, quality control checks are included and follow the workflow set out in the document "Quality Control and Data Recording for DDNS".
Before start
This protocol describes the amplification of the VP1 region, sample barcoding and library preparation. We anticipate users will have performed an RNA extraction prior to this protocol to extract Poliovirus RNA. We recommend the MagMAX Viral RNA Isolation Kit for this process.
Barcoded VP1 Primers:
To allow a simplified protocol, we use a 96-well primer plate with 5µM barcoded Y7 primer and 5µM barcoded Q8 primer in each well to give a total primer concentration of 10µM.
Each well contains Q8 and Y7 primers with the same unique barcode e.g A1 = Y7 with barcode 1 and Q8 with barcode 1, A2 = Y7 with barcode 2 and Q8 with barcode 2, etc.
The full set of 96 barcoded primer sequences are shown in Dataset_S1 of Shaw et al, 2020 and in the attached spreadsheet. BarcodedPrimers.xlsx
Steps
Sample Organisation
Pairs of samples (with the same EPID) can have consecutive barcodes but try not to group samples from the same geographic area together. This helps detect any potential cross-contamination because identical sequences are then unlikely to be detected in samples with consecutive barcodes that are adjacent to one another on the 96-well plate.
Record sample data, and the order for the samples in your csv file. At this point you can also add any other metadata that you have for the samples.
Here is an example of a barcode csv file: barcodes.csv
It is advised that you edit the name of the file so it is unique for each run you analyse.
You should also include any positive and negative controls in your list of samples. A positive control (resuspended Coxsackievirus A20 provided by NIBSC) and negative control (water) should each be included on the first and last RNA extraction batches of the day at least.
If any samples are repeats from a previous run, note this down in the appropriate column.
If there has been a delay in the processing of the sample e.g. due to a lack of extraction kits or software updates preventing the run, note "Yes" in the column "DelaysInProcessingForDDNS" and enter the type of delay in the column "DetailsOfDelays."
First Round PCR (semi-nest)
Prepare a master mix using the reaction volumes detailed in the table below for the number of samples you have plus negative controls. The reaction mix and SSIII enzyme are provided in
Forward primer: Y7 [GGGTTTGTGTCAGCCTGTAATGA]
Reverse Primers: Cre [TCAATACGGTGTTTGCTCTTGAACTG] (Arita et al. 2015)
nOPV-MM-R [TCGATACGGTGCTTGGATTTAAATTG]
| A | B |
|---|---|
| Reagent | 1 reaction (μL) |
| 2x Reaction mix | 12.5 |
| SSIII Platinum Taq mix | 1 |
| Reverse primers (10μM) | 1 |
| Nuclease free water | 4.5 |
Table1: Mastermix contents for a single first round PCR reaction. This can be multiplied up to fit the number of reactions you will be carrying out.
Vortex the mastermix for 3 seconds and spin down for 5 seconds to gather contents at the bottom of the tube. Aliquot 19μL to each PCR tube and add 5μL of sample RNA or nuclease free water for negative controls.
Incubate in a thermocycler for 30 minutes at 50°C.
Add 1μL of the forward primer to each reaction.
Amplify using the following cycling conditions:
| A | B | C | D |
|---|---|---|---|
| Cycle | Step | Temperature (°C) | Time |
| 1 | Initial denaturation | 94 | 2 minutes |
| 42 | Denaturation | 94 | 15 seconds |
| Annealing | 55 | 30 seconds | |
| Extension | 68 | 2 minutes 30 seconds | |
| 1 | Final extension | 68 | 5 minutes |
| - | Hold | 10 | - |
Table 2: Cycling conditions for the first round PCR
Once the PCR is finished, check to see if any reactions have evaporated, if so note this down in the sample csv.
Second Round PCR (VP1 amplification)
VP1 amplification is performed using barcoded primers as described in Dataset_S1 in Shaw et al 2020. These should be ordered in a 96-well plate layout and the forward and reverse primers premixed to make an overall 10μM working stock.
Prepare a mastermix as described below using
| A | B |
|---|---|
| Reagent | 1 Reaction (μL) |
| DreamTaq 2x mastermix | 12.5 |
| Nuclease free water | 8.5 |
Table 3: Mastermix contents for second round PCR using barcoded primers.
Vortex the mastermix for 3 seconds and spin down for 5 seconds to gather contents at the bottom of the tube.
Aliquot 21μL for each well of a 96-well PCR plate and add 2μL of 10uM barcoded primers (ensuring a different barcode is used for each sample) and 2μL of first round PCR product or nuclease free water for PCR negative controls.
Amplify using the following cycling conditions:
| A | B | C | D |
|---|---|---|---|
| Cycle | Step | Temp (C) | Time |
| 1 | Initial Denaturation | 95 | 2 minutes |
| 35 | Denaturation | 95 | 30 seconds |
| Annealing | 55 | 30 seconds | |
| Extension | 72 | 1 minute | |
| 1 | Final Extension | 72 | 10 minutes |
| - | Hold | 10 |
Table 4: Cycling conditions for VP1 PCR
Check all positive and negative controls from the VP1 reaction on a 1% agarose gel. The expected band for the positive control is around 1.2kb.
All samples can be marked as “Pass” for the PositiveControlCheck if all positive controls extracted on the same day show a VP1 band on the gel.
All samples can be marked as “Pass” for the NegativeControlCheck if all negative controls extracted on the same day show no VP1 band on the gel.
If any positive controls fail, or any negative controls have a band, all samples must be marked as fail.
If the positive control check is failed, run the positive control first round PCR product(s) on a 1% gel.
If there is no band, repeat the nested VP1 reaction for the control. If a band is visible, discard the VP1 amplicons and repeat the VP1 reactions for all samples.
If there is no band visible after repeating the nested VP1 reaction, repeat the RNA extractions after checking the RNA extraction kit is being used correctly and has not expired.
If the negative control check is failed, repeat both the first round PCR and the nested VP1.
If the negative control still shows a band on a gel or tapestation:
- Thoroughly clean the PCR and RNA extraction workstations.
- Replace each of the First Round and VP1 reagents in turn whilst performing blank reactions to determine a contaminated reagent.
- Perform an additional Negative RNA extraction to confirm that that RNA extraction kit is not contaminated.
Library Preparation for ONT MinION: Pooling, End-prep, and Adapter ligation
Pool 2μL of each VP1 PCR product into a 1.5mL tube and concentrate with AMPure beads
Add a volume of AMPure beads equal to the volume of the pooled VP1 products and incubate at room temperature for 5 minutes. Flick gently after 2 minutes to aid binding.
e.g. 50 samples, 2µl each pooled = 100µl pool, so add 100ul AMPure beads
Spin down the tube for 3 seconds then place on a magnetic rack until all the beads have formed a pellet and the solution is clear.
Pipette off the solution, avoiding disturbing the bead pellet.
Add 200μL of 80% Ethanol to the tube, leave for 30seconds, then remove and discard.
Repeat.
Spin down the tube for 2 seconds, place back on the magnet, then remove any remaining Ethanol.
Allow the pellet to air dry for 30 seconds or until dry but not cracking
Take the tube off the magnet and add 51μL of nuclease free water. Flick the tube to resuspend the beads and incubate at room temperature for 2 minutes.
Spin down the tube for 3 seconds then place back on the magnet, allowing the beads to pellet completely.
Remove 50μL of the eluted DNA and add to a clean 0.2mL PCR tube.
End-preparation:
Add the following reagents from
| A | B |
|---|---|
| Component | Volume (μL) |
| UltraII End-prep reaction buffer | 7 |
| UltraII End-prep enzyme mix | 3 |
Table 5: Reaction for end-prep of your pooled library
Mix gently by flicking the tube and spin down for 3 seconds.
In the thermocyler, incubate for 5 minutes at 20°C followed by 5 minutes at 65°C
Transfer to a 1.5mL tube and perform an AMPure bead clean.
Vortex the AMPure beads until all the beads are well mixed.
Add 60μl of resuspended beads to the tube and flick the tube to mix.
Incubate at room temperature for 5 minutes. Flick gently after 2 minutes to aid binding.
Spin down the tube for 3 seconds then place on a magnetic rack until all the beads have formed a pellet and the solution is clear.
Pipette off the solution, avoiding disturbing the bead pellet.
Add 200μL of 80% Ethanol to the tube, leave for 30seconds, then remove and discard.
Repeat.
Spin down the tube for 2 seconds, place back on the magnet, then remove any remaining Ethanol.
Allow the pellet to air dry for 1 minute or until dry but not cracking
Take the tube off the magnet and add 61μL of nuclease free water. Flick the tube to resuspend the beads and incubate at room temperature for 2 minutes.
Spin down the tube for 3 seconds then place back on the magnet, allowing the beads to pellet completely.
Remove 60μL of the eluted DNA and add to a clean 1.5mL tube.
From
Spin down the NEB Quick T4 Ligase and place on ice
From
Spin down and thaw Ligation Adapter (LA) on ice.
Thaw Ligation Buffer (LNB) at room temperature, spin down, mix by pipetting, then place on ice.
Thaw Elution Buffer (EB), and Short Fragment Buffer (SFB) at room temperature, mix by vortexing then place on ice.
If you plan to start the run on the same day, remove the Flush buffer, flush tether (FLT) and BSA from the freezer and thaw at room temperature. Once thawed, place on ice.
Remove your flow cell (FLO-MIN114, R10.4.1) from the fridge to allow it to get to room temperature.
Prepare the following reaction mix adding reagents to the 1.5mL tube with end-prepped DNA:
| A | B |
|---|---|
| Component | Volume (μL) |
| End-prepped DNA | 60 |
| Ligation buffer (LNB) | 25 |
| Quick T4 Ligase | 10 |
| Ligation Adapter (LA) | 5 |
Table6: Reaction mix for sequencing adapter ligation
Mix gently by flicking the tube then spin down.
Incubate at room temperature for 10 minutes.
During this time, you can run your flow cell check
Plug in your sequencing device, open the lid and insert your flowcell. In the MinKNOW software, navigate to the start panel then select flowcell check, then start. This will tell you how many pores are available for sequencing.
If your flow cell has been used before, instead of running a flow cell check, start a dummy sequencing run by selecting Start Sequencing, name the run "flowcell_check", select any kit, then set the time to 10 minutes, skip to final review then start run. At the beginning of the run it will do a short flow cell check and give a more accurate number for the available pores (calculated by adding together the available and unavailable pores in the pore status graph (see example image below)).
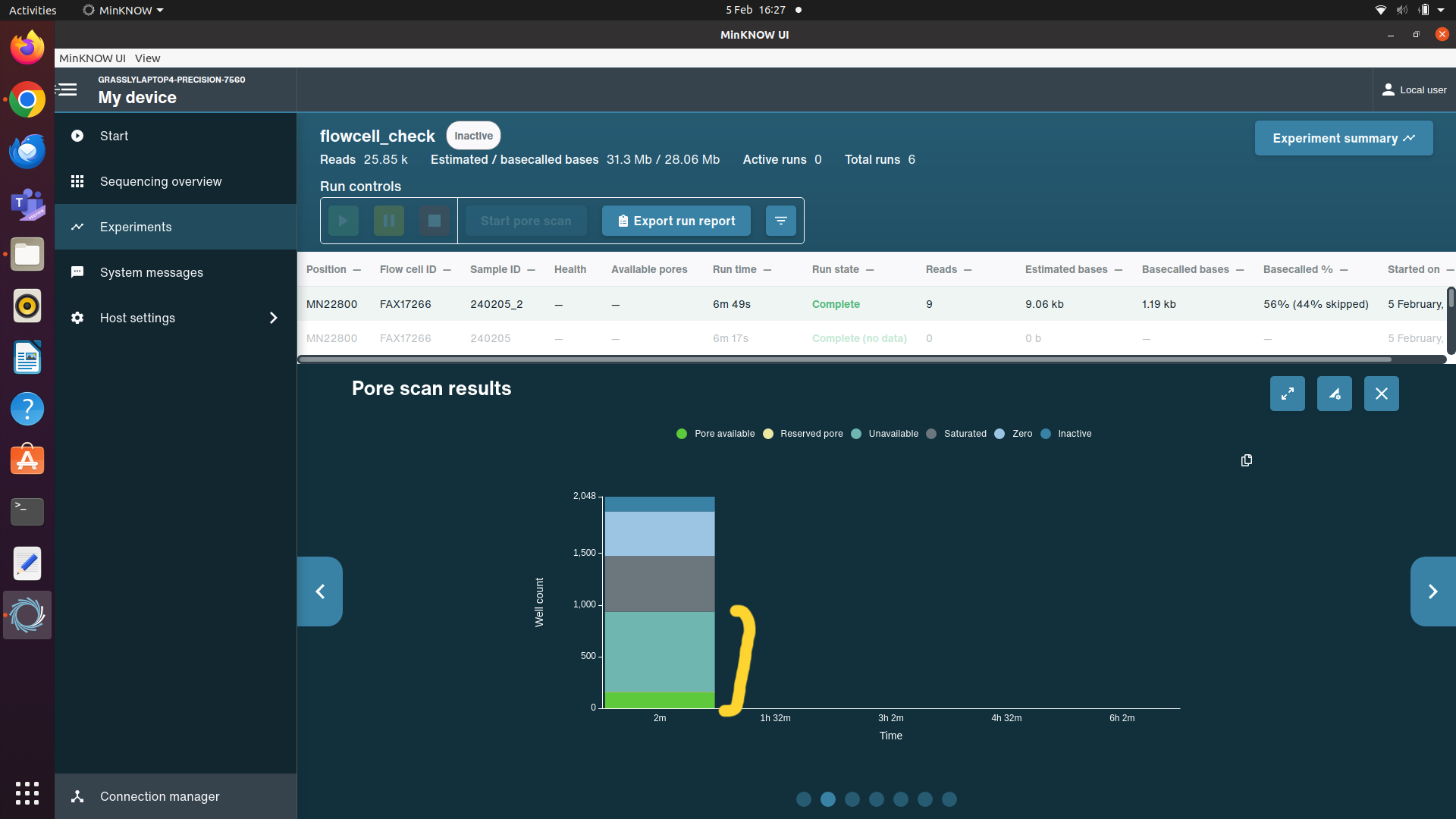
If a flow cell has less than 700 pores, do not use it for a 96 sample DDNS run. Take out a different flow cell and perform a flow cell check. The number of pores available in the flow cell you use should be noted down in the sample csv in the column “PoresAvilableAtFlowCellCheck” and the flow cell ID should be recorded in the “FlowCellID” column. Also record the number of times the flow cell has been used in the column “FlowCellUses”.
Carry out an AMPure bead purification using 40μL of resuspended AMPure XP beads.
Note: This clean-up is different to previous as it uses the ONT Short Fragment Buffer (SFB) and Elution buffer (EB) instead of 80% ethanol and water.
Vortex the AMPure beads until all the beads are well mixed.
Add 40μL of resuspended beads to the 1.5mL tube and mix by flicking the tube.
Incubate at room temperature for 5 minutes. Flick gently after 2 minutes to aid binding.
Spin down the tube for 3 seconds then place on a magnetic rack until all the beads have formed a pellet and the solution is clear.
Pipette off the solution, avoiding disturbing the bead pellet.
Add 250μL of Short Fragment Buffer (SFB). Remove the tube from the magnet and resuspend the beads in the SFB by flicking the tube.
Spin down for 3 seconds then return the tube to the magnet.
Allow the beads to pellet, then remove and discard.
Repeat.
Spin down the tube for 2 seconds, place back on the magnet, then remove any remaining SFB.
Allow the pellet to air dry for 1 minute or until dry but not cracked
Take the tube off the magnet and add 15μL of Elution buffer (EB). Flick gently to resuspend the beads and incubate at room temperature for 10 minutes.
Spin down the tube for 3 seconds then place back on the magnet, allowing the beads to pellet completely.
Remove 12µl of the eluted DNA and transfer to a clean 1.5ml tube.
Priming and Loading of the MinION Flowcell
Thaw the Sequencing buffer (SB), Library beads (LIB), Flow Cell Tether (FCT) and one tube of Flow Cell Flush (FCF) at room temperature then place on ice.
Mix the SQB, FCF, and FCT by vortexing, spin down, and return to ice. Spin down the LIB then place back on ice.
To create the priming mix the following reagents in a clean 1.5ml tube:
| A | B |
|---|---|
| Reagent | Volume (μL) |
| Flow cell flush (FCF) | 1,170 |
| Flow cell tether (FCT) | 30 |
| BSA (50mg/ml) | 5 |
Mix by pipetting and spin down. Place on ice until ready to use.
Open the lid of the nanopore sequencing device and slide the flow cell's priming port cover clockwise so that the priming port is visible. After opening the priming port, check for any bubbles under the cover. Draw back a small volume to remove any bubbles (a few µLs). Visually check that there is continuous buffer from the priming port across the sensor array.
Using a P1000 pipette, slowly load 800μL of the priming mix into the flow cell via the priming port.
Leave a small amount of liquid in the end of the pipette tip to ensure you do not introduce air into the flowcell.
Leave for 5 minutes.
Mix the contents of the LIB tube by pipetting just before adding to the following library mix in a 1.5ml tube:
| A | B |
|---|---|
| Reagent | Volume (μL) |
| DNA library | 12 |
| Sequencing buffer (SB) | 37.5 |
| Library beads (LIB) | 25.5 |
Complete the flowcell priming by opening the SpotOn port cover and carefully loading 200μL of the priming mix into the priming port. As before, leave a small amount of liquid in the bottom of the tip to avoid the introduction of air bubbles.
When adding the priming mix, you may see a small amount of liquid come up through the SpotOn port. If you do, pause and allow the liquid to flow back into the flowcell before continuing putting through the priming mix.
Mix the prepared library mix gently by pipetting.
Add the library mix to the flowcell via the SpotOn port in a dropwise fashion, allowing each drop to flow into the flowcell before adding the next.
Replace the SpotOn port cover and close the priming port, then replace the lid of your sequencing device.
Open the ONT MinKNOW software and follow the steps below to set up and start your sequencing run.
Click start, then start sequencing.
Create a name for you sequencing run, it is good practise to make this unique and identifiable for if you ever need to revisit the data. The date and an experiment name are recommended. In sample name you can put a number or repeat the experiment name - this is not as important as the run name. Then click continue.
Select the kit used - this is SQK-LSK114. Once you click this the barcoding options will appear. Select EXP-PBC096, then click continue
In the run length options, set the run time to 4 hours for a prospective DDNS run. Click continue.
In the basecalling options, select high accuracy basecalling. In the barcoding options, make sure barcoding is enabled and toggle to use barcode at both ends. Click continue until you reach the run overview, where you can double check the selected options, then click start run.
In your sample csv, record the run number in the column "RunNumber", the date in "DateSeqRunLoaded", and the run duration in "RunHoursDuration".
Washing your flow cell
After the sequencing run is finished you can wash your flow cell to remove the remaining library and either prepare for another sequencing run or for storage at 4°C . The wash uses the reagents supplied in the ONT wash kit:
Thaw the Wash Diluent at room temperature and mix briefly by vortexing.
Spin down the tube of wash mix (WMX) and place on ice.
Prepare the following wash solution in a clean 1.5ml tube
| A | B |
|---|---|
| Wash diluent | 398μL |
| Wash mix | 2μL |
Open the Priming port and using a P1000 pipette, carefully remove a small amount of liquid to remove any air bubbles under the port.
Carefully add 200μL of the wash solution through the priming port, leaving a small amount of liquid in the tip towards the end to avoid introducing an air bubble. Close the priming port and incubate for 5 minutes at room temperature.
Add the remaining 200μL of wash solution through the priming port. Close the priming port and incubate at room temperature for one hour.
At this point you can remove all waste from the waste channel, ensuring that the Priming Port is closed before doing so.
You can also put a label on the packaging of the flow cell to detail the date it was run, what was run on it, and for how long e.g. 13/03/2024 DDNS-run30 4 hours
If you will be storing the flow cell for future reuse, take out the bottle of Storage Buffer from the Wash Kit to thaw at room temperature.
After incubation you can either follow the flow cell priming and loading steps starting from Step 22 to load a new run, or the following steps for storing the flow cell for future use.
Briefly vortex the thawed Storage Buffer to mix, then add 500μL slowly through the Priming Port.
Close the priming port before removing all waste from the waste channel. Place the flow cell back in its plastic box and envelope, then store at 4°C
