Whole-cell proteomics and Analysis with or without nutrient stress by Tandem Mass Tagging-based proteomics V2
Sharan Swarup, J. Wade Harper wade_harper@hms.harvard.edu, Kelsey Hickey
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
The analysis of relative protein abundance has emerged as an important tool in cell biology. Typically, it is possible to quantify >8000 proteins under standard conditions. Tandem Mass Tags (TMT) are isobaric reagents that contain a set of isotopically distinct reporter ions, which can be used to quantify individual peptides in distinct samples through multiplexing(McAlister et al., 2014). Because the TMT analysis is performed in multiplexed format (up to 18 plex), it is possible to examine the effect of different perturbations (treatments, time courses, etc) on the total abundance of the proteome and include replicate samples as desired. This protocol is applicable to many different cell types, although the number of proteins quantified may differ, depending on the complexity of the proteomes in individual cell types. The small amount of protein needed (50-100 μg) makes application of this approach simple for many different types of cells. This protocol explicitly is used to examine the effects of nutrient stress on protein abundance in cell lines with or without autophagy.
Before start
WORKFLOW:

Attachments
Steps
Cell culture and nutrient stress treatments
Wild-type HEK293 (human embryonic kidney, fetus, ATCC CRL-1573, RRID: CVCL_0045) cells or the analogous cells lacking either ATG7 or FIP200 (see DOI: XXX) were grown in Dulbecco’s modified Eagle’s medium (DMEM, high glucose and pyruvate) supplemented with 10% fetal calf serum and maintained in a 5% CO2 incubator at 37oC. Cells were maintained at <80% confluency throughout the course of experiments.
DMEM was removed and cells were washed 3 times with PBS followed by resuspending cells in EBSS or DMEM lacking amino acids prepared according to H. Chino, et al Mol Cell 74 , 909-921 e906 (2019). The investigator can select the length of time for starvation but this is typically 10-18 hours.
Harvest, precipitation and digestion
For whole proteome analysis, 50µg is required for each replicate. Cells from step 2 are washed with PBS three times. Cells were lysed by in UREA denaturing buffer (8M Urea, 150mM NaCl, 50mM EPPS pH8.0, containing mammalian protease inhibitor cocktail (Sigma), and Phos-STOP) Cell lysates were collected by cell scrapers and sonicated on ice for 10 seconds.
Centrifugate suspensions at 13000rpm,4°C,0h 0m 0s for 0h 10m 0s and collect the supernatant.
Transfer quantified protein lysate concentration and 50µg for each sample to a clean 1.5 mL protein Lo-Bind Eppendorf tubes. Reduce lysates for 0h 20m 0s at Room temperature with 5millimolar (mM) , and alkylate cysteine residues with 20millimolar (mM)(Room temperature, 0h 30m 0s).
Extract protein content by methanol-chloroform precipitation and subsequent MeOH washes.
Add 4x volumes of MeOH and vortex.
Air dry (or speed-vac) protein pellet down to remove all traces of MeOH.
Add 1x volume of chloroform and vortex.
Add 3x volume of water and vortex.
Spin down at Room temperature for 0h 5m 0s at high speed.
Aspirate and discard the upper aqueous phase. Do not disturb the protein disc at the interface of the aqueous phase (top) and organic phase (bottom).
Add 4x volumes of MeOH and vortex.
Spin down at Room temperature for 0h 5m 0s at high speed.
Aspirate and discard supernatant. Do not disturb the protein pellet at the bottom of the tube.
Repeat MeOH wash.
Resuspend protein pellets 100µL of 200millimolar (mM) (8.5).
Digest samples at 37°C for 2h 0m 0s with endoproteinase Lys-C (Wako, Japan) at a 1/200 enzyme/protein ratio.
Digest with Trypsin (1:100) for 6h 0m 0sat 37°C.
TMT-Labeling of samples
Add 5µL to each sample. Solubilize TMT reagents are in ACN as per manufacturer’s instructions and 5µL is used for every 50µg. Performe TMT labeling in a final concentration of 20-25% ACN. Add 20µL to bring the reaction volume at 125 µL.The number of samples, and hence the number of individual TMT reagents, will depend upon the design of the experiment.
Incubate for 1h 0m 0sat 37Room temperature to label the samples.
Combine 1% of each labeled sample together in a tube, quench the reaction with 4µL for 0h 15m 0s at 37Room temperature, and dry down using the speed-vac. This combined sample is used to perform the ratio check to test labeling efficiency. The remaining amount of each sample can be stored in the freezer.
Stage tip for Ratio Check
Resuspend the dried sample in 100µL . Check to ensure that the pH of the sample is ~3 (or lower) using pH strip.
Make stage tip by placing 6-8 “cookies” of C-18 embedded membranes in 200µL.
Perform C-18 cleanup:
Equilibrate C-18 with 100µL .
Wash C-18 with 50µL .
Wash C-18 with 100µL .
Load sample on to C-18 to bind peptides.
Wash bound peptides on C-18 with 50µL .
Elute peptides off C-18 with 50µL into a mass spec vial.
Dry down eluted peptides in speed-vac.
Reconstitute peptides in 10µL.
Perform ratio check by analyzing the total amount of reporter ions present, as measured by mass spectrometry, for each TMT reporter ion channel.
Quench the entire volume of each sample using 8µL for 0h 15m 0s at 37Room temperature. Combine samples in 2 ml Eppendorf tube to equal amounts, based on normalization values you obtain from the ratio check.
Dry down labeled, combined sample in speed-vac.
Re-constitute the sample in 750µL.
SepPak clean-up (C18 solid-phase extraction (SPE))
Place SepPak column into vacuum slot on a vacuum manifold.
Fill SepPak with 1mL.
Start the vacuum, gently, try to ensure that the pressure gauge is below 10. This can be achieved by opening one of the valves gently. The fluid should pass through slowly, drop wise.
Fill SepPak with 1mL, Flow Liquid through.
Fill SepPak with 1mL , Flow Liquid through, repeat 2 more times.
Replace tube with 2 ml collection tube.
Add Peptides, Flow Liquid through (~ 750 µl).
Wash with 1mL, Flow Liquid through, 2 times.
Replace 15 ml collection tube with 2 ml collection tube.
Elute with 750µL. Since there is residual liquid left in the SepPak, ensure that all the liquid flows through SepPak.
Dry down in SpeedVac 37Room temperature 0h 15m 0s or 30°C.
Fractionation
Resuspend sample in 100µL 8.
Fractionate using pH reverse-phase HPLC:
Fractionate samples by high-pH reverse-phase HPLC (Agilent LC1260) into 96 fractions over a 1h 30m 0s run.
Fractions are run through an Aeris peptide XB-c18 column (Phenomenex; 250 mm x 3.6 mm), with mobile phase A containing 5 and 10millimolar (mM) 8 and phase B containing 90 and 10millimolar (mM) 8 (all inLC-MS grade H20).
Combine fractionated samples (either 12 or 24 fractions) in a non-continuous manner into individual 1.5 ml Eppendorf tube (see outline below form Paulo et al., 2016).
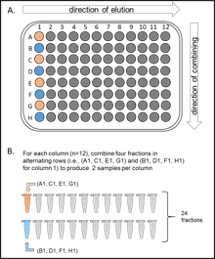
Dry down in SpeedVac.
Resuspend peptides in 100µL.
Check pH (~3.5) with pH indicator strips.
Stage tip for proteomics sample
Stage tip each fraction.
Make stage tips and equilibrate. Spin down at 3000rpm .
Perform C-18 cleanup:
Wash with 50µL.
Wash with 100µL .
Load sample
Collect flow through and freeze.
Wash with 50µL.
Elute with 50µL in mass spec vial.
Dry down in SpeedVac.
Reconstitute pellet in 10µL .
Freeze sample at --20°C until ready to run proteomics.
