Tn5-Duplex-Sequencing (Tn5-Duplex-Seq) for low-input single-molecule variant detection
Diane Shao, Nazia Hilal, Sangita Choudhury
Duplex-sequencing technology
Tn5-duplex-sequencing
Somatic mutations
single nucleotide variants (SNVs)
variation calling
copy number
variant allele frequency (VAF) analysis
Abstract
DNA mutations are the inevitable consequences of errors that arise during replication-repair of DNA damage as well as aging and disease progression. Because of their random and infrequent occurrence, quantification, and characterization of DNA mutations in the genome of somatic cells have been difficult. These mutations in DNA drive genetic diversity, alter gene function, define evolutionary trajectories, and provide targets for precision medicine and diagnostics. It is crucial to detect mutations across a wide range of abundance, i.e., variant allele frequency (VAF). Detecting low-abundance mutations (e.g., <0.1–1% VAF or in individual cells) is important for understanding human embryonic development, somatic mosaicism, and clonal hematopoiesis and uncovering pathogenic variants. Altogether somatic mutations provide important and unique insights into the biology of complex diseases. To decipher the causal inference, we must build robust genetic maps of somatic evolution in health and disease. The recent advent of duplex consensus sequencing has heralded a new generation of accuracy. However, multiple techniques focus on targeted areas of the genome (Twin Strand Biosciences) or are limited to restriction sites (Nanoseq), limiting their application to comprehensive somatic variant characterization. Furthermore, fragmentation of the genome and standard A-tailing and ligation creates errors (BotseqS, CODEC). Ligation of duplex strands for efficient sequencing has proven promising, though in practice requires complex molecular structures (Pro-Seq, CODEC) which have been observed to frequently result in incorrectly paired duplexes (CODEC). To enable comprehensive variant detection by next-generation DNA sequencing, we propose an innovative, accessible, and highly accurate Tn5 transposase-based duplex-sequencing technology ( Tn5-duplex-seq ) where complementary strands of DNA could be labeled at the molecular level in a single-tube reaction; thus, identifying single nucleotide variants (SNVs) from single-molecules of DNA A regardless of starting from single cells or pooled cell/DNA input. . The conceptual basis of the protocol comes from META-CS (Xing et al.2021), a Tn5 based aproach optimized for single-cell whole genome amplification. We find that modifications of this approach to include flexible input and the sequencing strategy to optimize cost per variant detection enables great flexibility for all low-input applications.
Tn5-duplex-seq approach offers several benefits over other duplex approaches including.
(1) preservation of original template molecules by utilizing 16 unique sequences (Compared to the loss of 50% of
molecules due to intramolecular symmetry during TN5-based Nextera library preparation)
(2) accuracy by eliminating the requirement for A-tailing
(3) efficiency of duplex capture through specifying input
(4) accessibility by using standard reagents and oligonucleotide preparations
(5) distinction between double-stranded SNVs and single-stranded lesions.
Our method enables library preparation for short-read sequencing. Downstream analysis enables accurate and high-throughput SNV/indel and copy number analysis.
Steps
PROTOCOL MATERIALS
100millimolar (mM)
10millimolar (mM)
20mg/mL
RECIPE FOR MAKING IN-HOUSE REAGENTS
2X Single Cell Lysis Buffer
40millimolar (mM)
40millimolar (mM)
0.3% volume
12X quenching solution
600millimolar (mM)
90millimolar (mM) 0.02% volume
- Reconstitute the 16 ADP1 and 16 ADP2 primers separately in low TE and store in aliquots at
-80°Cuntil ready for use. - Make an equimolar mix of the 16 ADP1 and ADP2 primers to make the ADP1 and ADP2 mix respectively.
TRANSPOSOME LOADING
Transposon Annealing
- Reconstitute 16 META-CS oligos and 1 reverse oligo to
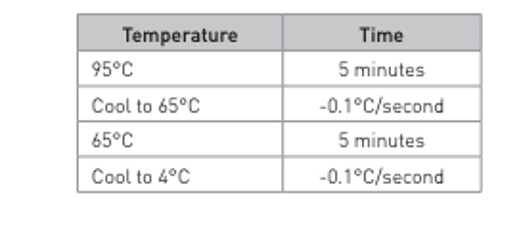
100micromolar (µM)in Annealing Buffer (40millimolar (mM)Tris-HCl (8),50millimolar (mM)NaCl) - Combine 1:1 of a singular META-CS oligo with the reverse oligo (there should be 16 separate reactions to put on the thermocycler). Mix up the reaction, spin it down briefly, and run the thermocycler using the conditions below:
Transposon Assembly.
- Combine all 16 reactions into one tube and aliquot for storage at
-80°C. - Take
10μLof this aliquot and combine it with 10μL of unloaded Transposome - Incubate at
23°Cfor0h 30m 0s - Add
10μLof 100% glycerol. - Aliquot and store at
-80°C.NoteEstimated final concentration including glycerol storage (~16.7 μM dimerized Tn5) *Prior to use, dilute Tn5 in Diagenode Tn5 dilution buffer depending on the desired concentration Optimization of Tn5 concentration: Check on 50 cells using dilutions of 1:500, 1:750, 1:1000, and 1:1500, and check the tagmentation curve.
TN5-DUPLEX LIBRARY PROCEDURE
Sorting and lysing cells 2μL
- Prepare nuclei for sorting.
- Sort cells directly into
2μLof 1X cell lysis buffer Run the thermocycler using the conditions below65°C
for for hold The plate can be stored after lysis.
Tn5 tagmentation 8μL
- Add
8μLtransposition mix (total10μLreaction). Vortex, spin down. 5μLDiagenode 2X Tagmentation buffer1μLdiluted Tn5 per optimized dilution instructions above2μLH2O Incubate in thermocycler using the conditions below65°C
for hold
Quenching 2μL
- Prepare 6X Stop Mix and add
2μLMix per tube. Spin down, vortex, and spin down. - Incubate in thermocycler using the conditions below
65°C
for for hold
First Strand tagging 13μL
Add 13μL Strand Tagging Mix 1. Vortex and spin down.
5μLQ5 Reaction Buffer5μLμL Q5 High GC Enhancer0.6μL100millimolar (mM)MgCl20.5μL10millimolar (mM)dNTP mix0.25μLBSA 20mg/ml0.25μLQ5 polymerase0.85μL``100micromolar (µM)ADP1 primer mix0.55μLH2O Incubate in thermocycler using the conditions below105°C
for
for for for hold
Stop reaction 1μL
Add 1μL Thermolabile ExoI per tube. Try to touch the minimum of the solution surface. Spin down first, then plate mix, and spin down again.
for for hold
Second Strand tagging 4μL
Add 4μL Strand Tagging 2 Mix (total 30μL). Vortex and spin down.
1μLQ5 Reaction Buffer1μLQ5 High GC Enhancer0.95μL100micromolar (µM)ADP2 primer mix0.1μL10millimolar (mM)each dNTP mix0.1μLQ5 polymerase0.85μLH2O Incubate in thermocycler using the conditions below105°C.
for for for hold
Stop reaction 1μL
Add 1μL Thermolabile ExoI per tube. Try to touch the minimum of the solution surface. Spin down first, then plate mix, and spin down again.
for 37°C for 0h 15m 0s for 65°C for0h 5m 0s hold
Library prep 14μL
- Make PCR Mix (per cell):
5μLNEB Universal Primer (NEB E7335S, E7500S, E7710S, E7730S)4μLQ5 Reaction Buffer4μLQ5 High GC Enhancer0.4μL10millimolar (mM)each dNTP mix0.4μLwater (H2O)0.2μLQ5 polymerase *add last 2.Add5μLNEB Index Primer per tube, avoiding touching the liquid.
3.Add 14μL PCR Mix per tube, avoiding touching the liquid. Vortex and spin down.
- Incubate in thermocycler using the conditions below
for 12 cycles of for , for for hold
PURIFICATION AND ZYMO CLEAN
Zymo clean
- Utilize the Zymo DNA Clean & Concentrator Kit with associated protocol (abbreviated version below).
- For microbulk samples, use 4:1 DNA binding buffer to sample (
200μLbuffer to50μLreaction). For single cell samples, pool desired samples first, then measure the total pooled volume and use 4:1 DNA binding buffer to sample volume. For single cell samples, pool 5 cells per spin column. For 50 cell samples, use 1 spin column per sample. - Add it to the spin column. The maximum volume that the spin column can hold is
800μLso pooled samples should have to be run through the same column sequentially until all of the liquid has been run through, discarding flowthrough each time. - Spin for
0h 0m 30sat maximum speed on the tabletop centrifuge >10,000xg at RT - Add
200μLwash buffer (with ethanol added) and centrifuge column - Repeat wash again
- Add
42μLx0.1 TE to elute and wait0h 4m 0sat room temperature - Spin for
0h 0m 30sat maximum speed on the tabletop centrifuge >10,000xg at RT
- Run
2μLon High Sensitivity D5000 TapeStation chip.
Size Selection (AMPure) 40μL
- Add (0.55X) resuspended AMPure XP beads to DNA library
22μL(0.55X) resuspended AMPure XP beads to40μLDNA library. Vortex and spin down. Label the tube as “A”. Incubate for0h 5m 0sat RT. - Place tube A on a magnetic stand for
0h 5m 0s. Carefully transfer the supernatant to a new tube. Label the new tube as “B”. - Size select tube “A” (0.55x AMPure XP beads):
a. Add
200μLof 80% freshly prepared ethanol to all tubes while in the magnetic stand, then carefully remove and discard the supernatant.
b. repeat the ethanol wash step one more time.
c. Let air dry on magnetic stand for 0h 1m 0s at RT.
d. Remove the tubes from the magnetic stand. Elute DNA from beads with 12μL 0.1X TE (for
single cell pools) or18μL 0.1X TE (for 50 cell pools). Vortex and gently spin down, incubate for 3 min at RT.
e. Place the tubes on the magnetic stand for 0h 3m 0s. Transfer 18μL of supernatant to a clean tube.
- Size select tube "B" (0.8x AMPure XP beads):
a. Add AMPure XP beads to DNA solution in tube B (0.15X)
8μLAMPure XP beads to DNA solution in tube B (0.15X). Vortex
and gently spin down. Incubate for0h 5m 0s at RT.
b. Place tube B on the magnetic stand for 0h 5m 0s. Remove supernatant from tube B.
i. IMPORTANT: save the supernatant in case AmPure did not work! (the DNA will still be in the supernatant)
c. Add 200μL of 80% freshly prepared ethanol to all tubes while in the magnetic stand, then carefully remove and discard the supernatant.
d. repeat the ethanol wash step one more time.
e. Let air dry on magnetic stand for 1 min at RT.
f. Remove the tubes from the magnetic stand. Elute DNA from beads with 12μL 0.1X TE (for
single cell pools) or18μL 0.1X TE (for 50 cell pools). Vortex and gently spin down, incubate for 3 min at RT.
g. Place the tubes on the magnetic stand for 0h 3m 0s. Transfer 18μL of supernatant to a clean tube.
- Run
2μLon High Sensitivity D5000 TapeStation chip. (Run1μLof sample and 1 of 0.1x TE instead of2μLof sample if you need to preserve sample). (Expected concentration for 5 single cell pools is:300-1000pg/μL. Expected concentration for 50 cell pools is:2000-15000pg/μL. - Proceed to DNA quantification with final product and dilute accordingly for sequencing.
Run TapeStation, need at least
5nanomolar (nM)concentration for sequencing. *Fraction B yields best sequencing results.
<img src="https://static.yanyin.tech/literature_test/protocol_io_true/protocols.io.6qpvr3nbzvmk/pp6dcb2up0_update.jpg" alt="-
Tube "A" (0.55x) contains DNA fragments with an average of 1000bp. Tube "B" (0.15x) contains DNA fragments with an average of 400-500bp (fragment size may range from 300bp-600bp). " loading="lazy" title="-
Tube "A" (0.55x) contains DNA fragments with an average of 1000bp. Tube "B" (0.15x) contains DNA fragments with an average of 400-500bp (fragment size may range from 300bp-600bp). "/>
Appendix-1
SI Appendix Table S1: Oligonucleotide sequences of META-CS transposon DNA and primers.
All oligos are HPLC purified
| A | B | C |
|---|---|---|
| A | B | |
| META transposon sequences: | ||
| META-CS-1 | GGCACCGAAAAAGATGTGTATAAGAGACAG | |
| META-CS-2 | CTCGGCGATAAAAGATGTGTATAAGAGACAG | |
| META-CS-3 | GGTGGAGCATAAAGATGTGTATAAGAGACAG | |
| META-CS-4 | CGAGCGCATTAAAGATGTGTATAAGAGACAG | |
| META-CS-5 | AGCCCGGTTATAAGATGTGTATAAGAGACAG | |
| META-CS-6 | TCGGCACCAATAAGATGTGTATAAGAGACAG | |
| META-CS-7 | GCCTGTGGATTAAGATGTGTATAAGAGACAG | |
| META-CS-8 | GCGACCCTTTTAAGATGTGTATAAGAGACAG | |
| META-CS-9 | GCATGCGGTAATAGATGTGTATAAGAGACAG | |
| META-CS-10 | GCGTTGCCATATAGATGTGTATAAGAGACAG | |
| META-CS-11 | GGCCGCATTTATAGATGTGTATAAGAGACAG | |
| META-CS-12 | ACCGCCTCTATTAGATGTGTATAAGAGACAG | |
| META-CS-13 | CCGTGCCAAAATAGATGTGTATAAGAGACAG | |
| META-CS-14 | TCTCCGGGAATTAGATGTGTATAAGAGACAG | |
| META-CS-15 | CCGCGCTTATTTAGATGTGTATAAGAGACAG | |
| META-CS-16 | CTGAGCTCGTTTTAGATGTGTATAAGAGACAG | |
| META-CS-rev | /5Phos/CTGTCTCTTATACACATC/3InvdT/ | |
| Adp1 primer mix: | ||
| META-CS-1-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGGCACCGAAAAAGATGTGTATAAG | |
| META-CS-2-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTCGGCGATAAAAGATGTGTATAAG | |
| META-CS-3-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGGTGGAGCATAAAGATGTGTATAAG | |
| META-CS-4-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCGAGCGCATTAAAGATGTGTATAAG | |
| META-CS-5-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGCCCGGTTATAAGATGTGTATAAG | |
| META-CS-6-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCGGCACCAATAAGATGTGTATAAG | |
| META-CS-7-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCCTGTGGATTAAGATGTGTATAAG | |
| META-CS-8-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCGACCCTTTTAAGATGTGTATAAG | |
| META-CS-9-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCATGCGGTAATAGATGTGTATAAG | |
| META-CS-10-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCGTTGCCATATAGATGTGTATAAG | |
| META-CS-11-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGGCCGCATTTATAGATGTGTATAAG | |
| META-CS-12-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTACCGCCTCTATTAGATGTGTATAAG | |
| META-CS-13-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCCGTGCCAAAATAGATGTGTATAAG | |
| META-CS-14-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCTCCGGGAATTAGATGTGTATAAG | |
| META-CS-15-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCCGCGCTTATTTAGATGTGTATAAG | |
| META-CS-16-adp1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTGAGCTCGTTTTAGATGTGTATAAG | |
| Adp2 primer mix: | ||
| META-CS-1-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGCACCGAAAAAGATGTGTATAAG | |
| META-CS-2-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCGGCGATAAAAGATGTGTATAAG | |
| META-CS-3-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGTGGAGCATAAAGATGTGTATAAG | |
| META-CS-4-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCGAGCGCATTAAAGATGTGTATAAG | |
| META-CS-5-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGCCCGGTTATAAGATGTGTATAAG | |
| META-CS-6-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCGGCACCAATAAGATGTGTATAAG | |
| META-CS-7-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCCTGTGGATTAAGATGTGTATAAG | |
| META-CS-8-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCGACCCTTTTAAGATGTGTATAAG | |
| META-CS-9-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCATGCGGTAATAGATGTGTATAAG | |
| META-CS-10-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCGTTGCCATATAGATGTGTATAAG | |
| META-CS-11-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGCCGCATTTATAGATGTGTATAAG | |
| META-CS-12-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTACCGCCTCTATTAGATGTGTATAAG | |
| META-CS-13-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCCGTGCCAAAATAGATGTGTATAAG | |
| META-CS-14-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCTCCGGGAATTAGATGTGTATAAG | |
| META-CS-15-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCCGCGCTTATTTAGATGTGTATAAG | |
| META-CS-16-adp2 | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGAGCTCGTTTTAGATGTGTATAAG |
Indexes
| A | B |
|---|---|
| A | B |
| I7_Index_ID | NEB index primer |
| 1 | ATCACG |
| 2 | CGATGT |
| 3 | TTAGGC |
| 4 | TGACCA |
| 5 | ACAGTG |
| 6 | GCCAAT |
| 7 | CAGATC |
| 8 | ACTTGA |
| 9 | GATCAG |
| 10 | TAGCTT |
| 11 | GGCTAC |
| 12 | CTTGTA |
| 13 | AGTCAA |
| 14 | AGTTCC |
| 15 | ATGTCA |
| 16 | CCGTCC |
| 17 | GTAGAG |
| 18 | GTCCGC |
| 19 | GTGAAA |
| 20 | GTGGCC |
| 21 | GTTTCG |
| 22 | CGTACG |
| 23 | GAGTGG |
| 24 | GGTAGC |
| 25 | ACTGAT |
| 26 | ATGAGC |
| 27 | ATTCCT |
| 28 | CAAAAG |
| 29 | CAACTA |
| 30 | CACCGG |
| 31 | CACGAT |
| 32 | CACTCA |
| 33 | CAGGCG |
| 34 | CATGGC |
| 35 | CATTTT |
| 36 | CCAACA |
| 37 | CGGAAT |
| 38 | CTAGCT |
| 39 | CTATAC |
| 40 | GTGATC |
| 41 | GACGAC |
| 42 | TAATCG |
| 43 | TACAGC |
| 44 | TATAAT |
| 45 | TCATTC |
| 46 | TCCCGA |
| 47 | TCGAAG |
| 48 | TCGGCA |
