Plasmid Sequence Analysis from Long Reads
David A Eccles
Abstract
This protocol demonstrates how to assemble reads from plasmid DNA, and generate a circularised and non-repetitive consensus sequence
At the moment, this protocol uses Canu to de-novo assemble high-quality single-cut reads.
Input(s) :
- demultiplexed fastq files (see protocol Demultiplexing Nanopore reads with LAST). I've noticed that the default demultiplexing carried out by Guppy (at least up to v4.2.2, as used in the first version of this protocol) has issues with chimeric reads, which can affect assembly.
Output(s):
- Consensus sequence per barcode as a fasta file
Before start
When preparing plasmid DNA for sequencing, I follow one piece of advice that Dr. Divya Mirrington (ONT) gave me about pooling: create a pooled sample with volumes that you're confident about, then remove an aliquot from that for adding the remaining reagents [paraphrased]. I don't do any additional cleanup for purified plasmid DNA; they tend to sequence very well on flow cells without that cleanup.
My key requirement for plasmid sequencing is a concentration of >20 ng/μl (ideally by qubit or quantus). Concentrations over 100 ng/μl should be diluted down. If the plasmids can be diluted down to all exactly the same concentration (but at least 20 ng/μl), or they're all similar lengths, then that makes creating an equimolar pool much easier.
When creating the pools, I add at least 1 μl of the the sample that I need to add the least for (might be more if the total volume is less than 11 μl), then add the corresponding amount of other samples to create an equimolar pool. I then take 11 μl from the pool to be used for rapid adapter addition.
If samples are equal concentration:
Add amounts according to the length of the plasmid divided by the length of the shortest plasmid. For example, if there are two plasmids, one with length 3kb and another with length 35 kb, then add 1 μl of the 3kb plasmid, and 35/3 = 11.7 μl of the 35kb plasmid.
If plasmids are roughly equal length (i.e. less than ~10% length difference between plasmids):
Add amounts according to the concentration of the highest-concentration sample divided by the concentration of the plasmid. For example, if there are three plasmids, one with concentration 50 ng/μl, one with concentration 35 ng/μl, and one with concentration 20 ng/μl, then add 1 μl of the 50 ng/μl plasmid, 50/35 = 1.4 μl of the 35 ng/μl plasmid, and 2.5 μl of the 20 ng/μl plasmid. The total volume of this pool will be less than 11 μl (1 + 1.4 + 2.5 = 4.9 μl), so in this case I would triple these volumes (3 μl; 4.2 μl; 7.5 μl) to create a pool of > 11 μl.
If samples are different concentrations and different lengths:
Make the sample prep easier. Use multiple flow cells for different plasmid length ranges. Dilute higher-concentration samples down to the lowest-concentration samples. I don't recommend trying to do both calculations at the same time to determine added volumes because there's a much higher chance of getting added amounts wrong, leading to wasted samples or wasted flow cells.
If you have a sufficiently-accurate pipetting robot, a sample sheet, and someone who is comfortable with equations:
Pre-calculate amount to add assuming 12 μl total pool volume:
ratio = length / max( length ) * max( conc ) / conc
volume = ratio * 12 / sum( ratio )
[That's my guess at the right equations; please let me know if there's an error]
Steps
Read file preparation
Demultiplex reads as per protocol Demultiplexing Nanopore reads with LAST.
If this has been done, then the following command should produce output without errors:
for bc in $(awk '{print $2}' barcode_counts.txt);
do ls demultiplexed/reads_${bc}.fq.gz;
done
Example output:
demultiplexed/reads_BC02.fq.gz
demultiplexed/reads_BC03.fq.gz
demultiplexed/reads_BC04.fq.gz
demultiplexed/reads_BC05.fq.gz
demultiplexed/reads_BC07.fq.gz
demultiplexed/reads_BC09.fq.gz
If the barcode_counts.txt file is missing, the output will look like this:
awk: fatal: cannot open file `barcode_counts.txt' for reading (No such file or directory)
If one or more of the barcode-demultiplexed files are missing, the output will look something like this:
demultiplexed/reads_BC02.fq.gz
demultiplexed/reads_BC03.fq.gz
demultiplexed/reads_BC04.fq.gz
ls: cannot access 'demultiplexed/reads_BC05.fq.gz': No such file or directory
ls: cannot access 'demultiplexed/reads_BC07.fq.gz': No such file or directory
demultiplexed/reads_BC09.fq.gz
If reads have been demultiplexed by MinKNOW, then the following approach should work to create the right input format:
mkdir demultiplexed;
# readlocation is the directory that contains barcode subdirectories
readLocation = "../*/fastq_pass"; # or "../*/pass" for Guppy
for x in $(ls ${readLocation}); do
echo ${x};
cat ${readLocation}/${x}/*.fastq | gzip > demultiplexed/reads_${x}.fq.gz;
echo "1 ${x}" >> barcode_counts.txt;
done
Create a directory to store results files
mkdir results
Determine the N50/L50 read length for each barcode. This will be used as the initial guess at the assembly size.
(for bc in $(awk '{print $2}' barcode_counts.txt);
do echo -n ${bc};
fastx-length.pl demultiplexed/reads_${bc}.fq.gz 2>&1 > /dev/null | \
grep L50 | awk '{print "\t"$5$6}' | perl -pe 's/b$//';
done) > results/read_L50.txt
```This file can be viewed to confirm the assembly lengths:
cat results/read_L50.txt
BC02 347 BC03 8.904k BC04 8.888k BC05 10.262k BC07 11.076k BC09 11.093k
Read filitering
Filter out any reads that are less than half of the target read length, and determine the average quality of the remainder, keeping information on (at most) 100 of the highest-quality reads:
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
~/scripts/fastx-compstats.pl demultiplexed/reads_${bc}.fq.gz | \
sort -t ',' -k 16rn,16 | \
awk -F ',' -v "len=${len}" \
'BEGIN{if(match(len, "k") > 0){sub("k","",len); len=len*1000}}
{if($18 > (len / 2)){print}}' | \
head -n 100 | grep -v '^name' > results/bestLong_100X_${bc}.csv;
done
```Check how successful the sequencer was at getting 100X coverage by counting lines:
wc -l results/bestLong_100X_*.csv
90 results/bestLong_100X_BC02.csv 100 results/bestLong_100X_BC03.csv 100 results/bestLong_100X_BC04.csv 100 results/bestLong_100X_BC05.csv 100 results/bestLong_100X_BC07.csv 100 results/bestLong_100X_BC09.csv 590 total
Subset the original read sets to only include the high-quality long reads:
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
~/scripts/fastx-fetch.pl -i results/bestLong_100X_${bc}.csv \
demultiplexed/reads_${bc}.fq.gz > results/bestLong_100X_${bc}.fastq; done
Choice 1: mapping to a reference sequence
This protocol assumes the reference sequence has a name 'refName', and can be found at 'reference/refName.fa'. Change 'refName' here in the likely event that the sequence / file has a different name, then export the variable REFNAME to make subsequent steps easier.
export REFNAME="refName"
Create a LAST index for the reference sequence:
lastdb -uNEAR -R01 reference/${REFNAME}.fa reference/${REFNAME}.fa
Prepare a substitution matrix for barcode mapping. I recommend doing this by training LAST, and incorporating quality scores into the trained matrix:
last-train --matsym -Q 1 -P 10 reference/${REFNAME}.fa \
results/bestLong_100X_BC*.fastq
Copy the last few lines from the output into a text file called _plasmid.mat_
Here is a matrix that I have created from plasmid reads called using the guppy super-accuracy basecaller, v5.0.13:
#last -Q 1
#last -t4.39812
#last -a 17
#last -A 18
#last -b 4
#last -B 5
#last -S 1
# score matrix (query letters = columns, reference letters = rows):
A C G T
A 6 -45 -34 -46
C -45 6 -46 -45
G -34 -46 6 -46
T -46 -45 -46 6
```[plasmid.mat](https://static.yanyin.tech/literature_test/protocol_io_true/protocols.io.by7bpzin/fquijpt62.mat)
This matrix has a moderate penalty for opening gaps (i.e. insertions and deletions), and a lower penalty for inserting them. Insertions are slightly less likely than deletions. It has a higher penalty for A/G transition variants, and a similar penalty for C/T transition variants as other substitution penalties.
Map reads to the reference and convert to BAM format using maf-convert and samtools :
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
lastal -j 7 -P 10 -p plasmid.mat reference/${REFNAME}.fa \
results/bestLong_100X_${bc}.fastq | last-split -n -m 0.99 | last-postmask | \
maf-convert sam | samtools view -h --reference reference/${REFNAME}.fa | \
samtools sort > results/${bc}_vs_${REFNAME}.bam
samtools index results/${bc}_vs_${REFNAME}.bam
done
Split reads into forward and reverse-mapped sequences, and tally up base counts at each location:
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
samtools view -b -F 0x10 results/${bc}_vs_${REFNAME}.bam | \
samtools mpileup --reference reference/${REFNAME}.fa -B -Q 0 - | \
~/scripts/readstomper.pl -c > results/fwd_stomped_${bc}_vs_${REFNAME}.csv
samtools view -b -f 0x10 results/${bc}_vs_${REFNAME}.bam | \
samtools mpileup --reference reference/${REFNAME}.fa -B -Q 0 - | \
~/scripts/readstomper.pl -c > results/rev_stomped_${bc}_vs_${REFNAME}.csv
done
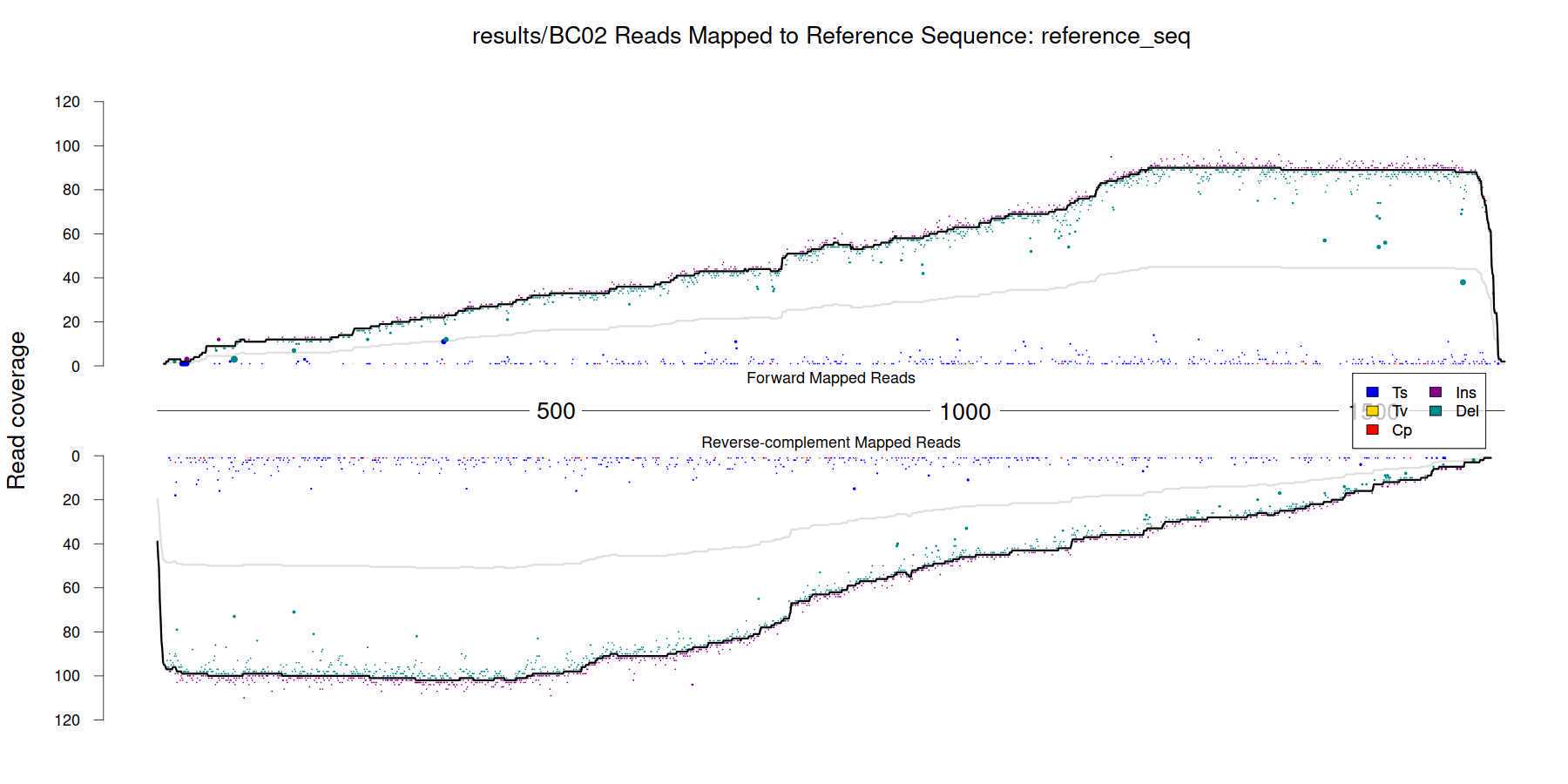
Create a visualisation of the base-stomped output:
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
~/scripts/stomp_plotter.r -f results/fwd_stomped_${bc}_vs_${REFNAME}.csv \
-r results/rev_stomped_BC02_vs_${REFNAME}.csv -prefix results/${bc}
done
Example output ( _results/BC02_stompPlot_reference_seq.png_ ):
Choice 2: De-novo Canu Assembly
Run a Canu assembly on each read set, using default options:
cat results/read_L50.txt | while read bc len;
do canu -nanopore results/bestLong_100X_${bc}.fastq \
-p ${bc} -d results/canu_${bc} genomeSize=${len};
done
Trim the assembled contigs based on Canu's trim recommendations:
cat results/read_L50.txt | while read bc len;
do echo ${bc} ${len};
samtools faidx results/canu_${bc}/${bc}.contigs.fasta \
$(grep '^>' results/canu_${bc}/${bc}.contigs.fasta | \
perl -pe 's/^>(.*?) .*trim=(.*)/$1:$2/') > results/circTrimmed_${bc}.fasta;
done
