Nano-CUT&Tag for multimodal profiling of the chromatin
Marek Bartosovic, Goncalo Castelo-Branco
Disclaimer
This protocol includes portions that are excerpted from works © 2018-2022 10x Genomics, Inc. (10x Genomics) and reproduced here with permission. All other rights reserved. The use of 10x Genomics products in practicing the methods set forth herein has not been validated by 10x Genomics, and such non-validated use is not covered by 10x Genomics’ standard warranty, and 10X GENOMICS HEREBY DISCLAIMS ANY AND ALL WARRANTIES FOR SUCH USE. Nothing in this document should be construed as altering, waiving or amending in any manner 10x Genomics’ terms and conditions of sale or use for its products and software, including without limitation such terms and conditions relating to certain use restrictions, limited license, warranty and limitation of liability, and nothing in this document shall be deemed to be Documentation, as that term is set forth in such terms and conditions of sale. Nothing in this document shall be construed as any representation by 10x Genomics that it currently or will at any time in the future offer or in any way support any application set forth herein.
Abstract
Nano-CUT&Tag is a multimodal technology to profile several histone modifications at the same with single-cell resolution. Nano-CUT&Tag implements a novel Tn5 fusion proteins to anti-mouse and anti-rabbit secondary nanobodies. Optionally, ATAC-seq can be performed prior on the same sample to profile open chromatin at the same time. Novel tagmentation protocol, which involves two-step tagmentation by MeA and MeB oligonucleotides yields increased number of fragments per cell comparing to previous single-cell CUT&Tag protocols on 10x Genomics platform.




Before start
Before starting, make yourself familiar with the 10x genomics Chromium Single Cell ATAC Reagent Kits User Guide (v1.1 Chemistry).
Follow all the best practices and tips given in the 10x genomics Chromium Single Cell ATAC Reagent Kits User Guide.
This protocol is compatible with Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 has not been tested with Chromium Next GEM Single Cell ATAC Kit v2.
CG000209_Chromium_NextGEM_SingleCell_ATAC_ReagentKits_v1.1_UserGuide_RevF.pdf
Steps
Tn5 loading
Annealing adaptor sequences:
Tn5_MeA_P5_noBCD.
5' TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG
Tn5_MeA_P5_bcdA.
5'-TCGTCGGCAGCGTCT TATAGCCT GCGATCGAGGACGGCAGATGTGTATAAGAGACAG
Tn5_MeA_P5_bcdB
5'-TCGTCGGCAGCGTCT ATAGAGGC GCGATCGAGGACGGCAGATGTGTATAAGAGACAG
Tn5_MeA_P5_bcdC
5'-TCGTCGGCAGCGTCT CCTATCCT GCGATCGAGGACGGCAGATGTGTATAAGAGACAG
Tn5ME-B:
5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′
Tn5MErev.
5'-[phos]CTGTCTCTTATACACATCT-3'
Resuspend the oligonucleotides (Tn5ME-A, Tn5ME-B, Tn5MErev) in water to a final concentration of 100µM each.
Mix equimolar amounts of Tn5MErev/Tn5ME-A and Tn5MErev/Tn5ME-B in separate 200 µl PCR tubes.
e.g:
tube1: 10µL Tn5ME-A + 10µL Tn5MErev
tube2: 10µL Tn5ME-B + 10µL Tn5MErev
Denature in the thermocycler for 0h 5m 0s at 95°C , and cool down slowly on the thermocycler by ramping down to 20°C by 0.1°C
Pause point: Store the annealed oligos at -20°C
Mix annealing mix.
The final Tn5 concentration is 2micromolar (µM) of Tn5 dimer
Use unique barcodes for specific nano-Tn5 or WT-Tn5.
Mouse Nano-Tn5 (MeA/P5 loaded):
4µL Annealed, barcoded MeA/Me-Rev oligos 50micromolar (µM) (e.g. barcode A, Me-A/Me-Rev)
21µL Glycerol
3µL Nano-Tn5 (mouse, 5mg/ml, MW = 73941 g/mol; 67.6micromolar (µM) )
22µL 2x Tn5 loading buffer
Rabbit Nano-Tn5 (MeA/P5 loaded):
4µL Annealed, barcoded MeA/Me-Rev oligos 50micromolar (µM) (e.g. barcode B, Me-A/Me-Rev)
21µL Glycerol
2.2µL Nano-Tn5 (rabbit, 6.8mg/ml, MW = 73013 g/mol 93.1micromolar (µM) )
22.8µL 2x 2x Tn5 loading buffer
WT Tn5-MeA with barcode (for ATAC-seq)
4µL Annealed, barcoded MeA/Me-Rev oligos 50micromolar (µM) (e.g. barcode C, Me-A/Me-Rev)
21µL Glycerol
3.1µL Tn5 (3.5 mg/ml, MW = 53300 g/mol 65.7micromolar (µM) )
21.9µL 2x 2x Tn5 loading buffer
WT Tn5-MeB unbarcoded (for 2nd tagmentation)
4µL Annealed MeB/Me-Rev oligos 50micromolar (µM) (un-barcoded, Me-B/Me-Rev)
21µL Glycerol
3.1µL Tn5 (3.5 mg/ml, MW = 53300 g/mol 65.7micromolar (µM) )
21.9µL 2x Tn5 loading buffer
2x Dialysis buffer
100 mM HEPES-KOH pH7.2, 200 mM NaCl, 0.2 mM EDTA, 0.2% Triton-X, 20% Glycerol, Store at 4°C ;
Add DTT fresh to the 2x dialysis buffer just before loading (2mM final). Keep 200 mM DTT stock at -20°C
For details on buffer preparation see Materials section
Incubate for 1h 0m 0s at Room temperature
Store the loaded nano-Tn5 at -20°C
ATAC-seq (optional)
Dissociate tissues/cells of interest by desired method and obtain single-cell suspension. Wash the cells once with 1x PBS
Centrifuge the cells for 0h 10m 0s at 300x g at 4°C . Discard the supernatant.
Add 200µL of ATAC lysis buffer (for 200,000 cells, see materials section for buffer recipe). Pipette up and down gently 3x and incubate on ice for 0h 3m 0s
Add 1mL of ATAC wash buffer and gently invert the tube 3x.
Centrifuge at 500x g for 0h 10m 0s . Discard the supernatant.
Prepare transposition mix:
100µL 2X TD Buffer (see materials section)
66µL 1X PBS2µL
10% Tween-20 (final 0.1% v/v2µL
1% Digitonin (final 0.01% v/`10µL`)
Tn5 Transposase (loaded with uniquely barcoded oligonucleotides-MeA onl`20µL`)
nuclease-free H2O
Resuspend the nuclei in 200µL of transposition mix (for 200,000 cells).
Incubate for 0h 30m 0s at 37°C in thermomixer at 1000 rpm.
Stop the tagmentation by adding 10µL of 500 mM EDTA. Mix by pipetting up and down 3x.
Centrifuge for 0h 10m 0s at 500x g
Remove the supernatant. Resuspend in 200µL of CUT&Tag Antibody buffer.
Centrifuge for 0h 3m 0s at 600x g. Remove the supernatant.
Proceed to CUT&Tag Antibody binding ( Step 23 ).
CUT&Tag Nuclei isolation (nano-CUT&Tag without ATAC)
Dissociate tissues/cells of interest by desired method and extract nuclei by incubation for 0h 3m 0s in 200µL of Antibody buffer on ice.
Centrifuge the nuclei for 0h 3m 0s at 600x g.
Remove the supernatant.



Antibody binding
Prepare antibody mix
Starting concentrations (can be further optimised, depending on the antibody)
1:100 primary antibody
1:100 nano-Tn5
Final volume 100ul per sample.
Resuspend the nuclei pelet in the prepared antibody mix (1:100 primary antibody, 1:100 nano-Tn5) by pipetting up and down 5x.
Incubate 0h 3m 0s on with rotation on a horizontal roller at 4°C

Washing and Tagmentation
The next day, centrifuge for 0h 3m 0s at 600xg.
Remove the supernatant and resuspend in 200 ul of Dig-300 wash buffer .
Repeat the steps 27-28 for total of 2 washes.
Resuspend the nuclei pellet in 200 ul of Tagmentation buffer. Pipette mix 5x to resuspend the pellet.
Incubate for 1h 0m 0s at 37°C
During the incubation prepare tagmentation STOP buffer.
Prepare 2x diluted nuclei buffer (DNB) from 20x nuclei buffer (10x scATAC-seq, PN: 2000207). Store the aliquots of 2x DNB at -20°C
Stop buffer:
100µL 2x Diluted Nuclei buffer
20µL 20% BSA
70µL water
10µL 500 mM EDTA).
1x DNB + BSA buffer:
500µL 2x Diluted Nuclei buffer (10x scATAC-seq kit, dilute down from 20x (PN: 2000207))
100µL 20% BSA
400µL water
Stop the tagmentation by removing from 37 thermoblock and adding 200µL of STOP buffer. Mix well by pipetting up and down 3x.
Centrifuge for 0h 3m 0s at 300x g. Remove supernatant
Resuspend the nuclei in 200µL of 1x DNB+BSA buffer.
Centrifuge for 0h 3m 0s at 300x g. Remove supernatant
Resuspend the nuclei in 200µL of 1x DNB+BSA buffer.
Centrifuge for 0h 3m 0s at 300x g.
Remove the most of the supernatant, leave the nuclei in cca 10µL of remaining 1xDNB+BSA buffer.
Add 10µL of 1xDNB+BSA buffer, for final 20 ul of nuclei suspension. Measure the exact volume using P20 pipette.
Nuclei counting )
Count the concentration of nuclei. Use 2µL of nuclei suspension and mix with 8µL of trypan blue. For counting use manual counting chambers (VWR, 630-1893). Do the counting in two replicates for more accuracy.

To aim for cca 5,000 recovered nuclei, load cca 16,000 nuclei or determine ratio of loaded/recovered nuclei empirically.
Chromium Next GEM barcoding
Mix the GEM generation and barcoding mix. Use the desired volume of nuclei and fill up to 15µL with 1x DNB+BSA buffer.
Prepare nuclei mix and keep it on ice:
7µL ATAC buffer B
XµL Nuclei (Use =< 8ul of nuclei)
up to 15µL 1x DNB+BSA buffer
keep on ice
Prepare barcoding mix and :
56.5µL Barcoding reagent B
1.5µL Reducing agent B
2µL Barcoding enzyme
keep it on ice
Assemble the Chromium Next GEM Chip H according to the manufacturer's instructions.

Load the Chromium Next GEM Chip H according to the manufacturer's instructions. Run the droplet generation.

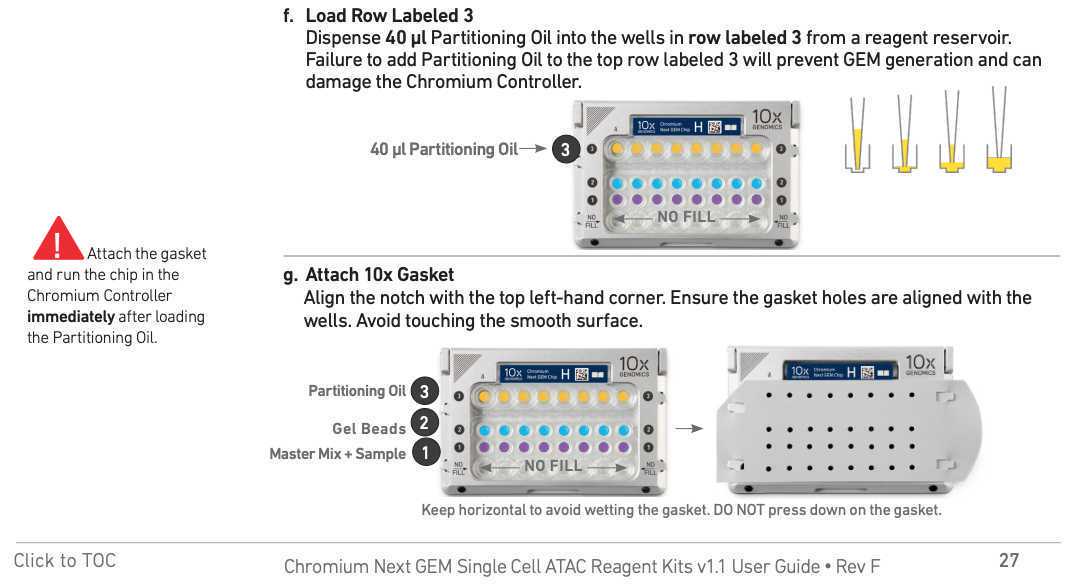
Recover droplets from the Next GEM Chip H according to the manufacturer's instructions.

Incubate in PCR cycler according to the manufacturer's instructions.
The linear amplification (LA) and single-cell barcoding occurs at this step.

Post-GEM incubation cleanup
Perform post-GEM incubation cleanup according to manufacturer's instructions - Steps 3.1-3.2
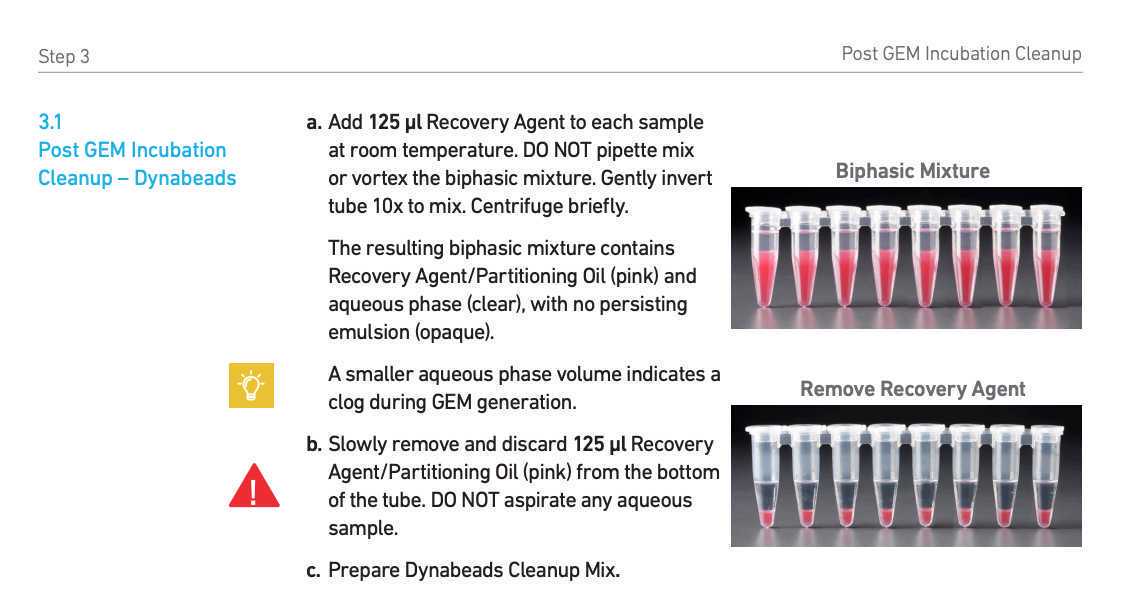
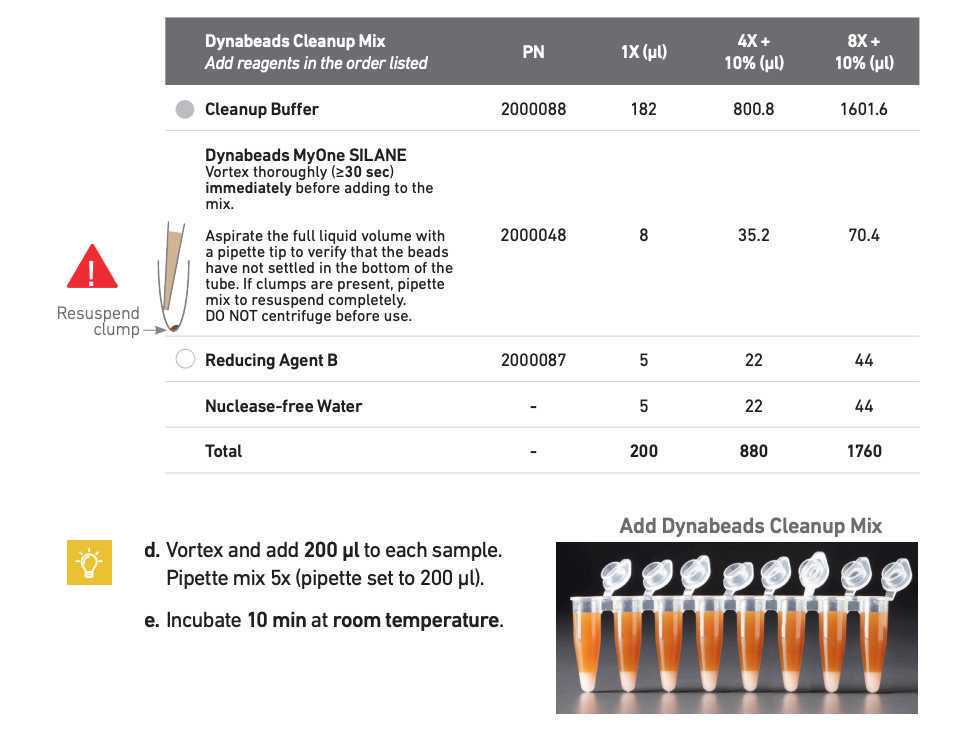


Use 2ul of the purified DNA to measure the concentration (optional) using Qubit high sensitivity dsDNA kit.
MeB tagmentation and library preparation
Mix MeB tagmentation reaction:
38-40µL Barcoded LA product (from previous step, typically 5-10 ng)
50µL 2x TD buffer
0.5µL MeB-loaded Tn5 (starting point, should be adjusted for specific batch of Tn5 and yield of DNA)
up to 100µL Water
Incubate in PCR cycler 0h 30m 0s at 37°C -> 4°C hold
Heated lid at 50°C
Purify the DNA using Zymo DNA Clean and concentrator-5 kit according to manufacturer's instructions.
Transfer the sample into 1.5 ml eppendorf tube.
Add 500µL of zymo binding buffer to your sample.
Wash 2x with 200µL of zymo wash buffer.
Perform one more dry spin 1min at max speed to remove residual liquid.
Elute the DNA in 40µL of DNA elution buffer (Zymo kit) . Incubate 2minutes on column, then centrifuge.
D4003T_D4003_D4004_D4013_D4014_DNA_Clean_Concentrator-5_ver_1_2_1_LKN-SW__1.pdf
Run PCR library amplification:
40µL Purified DNA from previous step (sample)
7.5µL SI-PCR primer B (10x ATAC-seq kit; PN: 2000128)
2.5µL Individual Single Index N Set A primer (product code: 1000212)
50µL AMP mix (10x ATAC-seq kit; PN: 2000047/ 2000103)
Incubate in PCR cycler with the following program:
Lid temperature 105°C , volume 100µL .
-
72°C5min -
98°C45sec -
98°C20sec | -
67°C30sec | Repeat 13x -
72°C20sec | -
72°C1min -
4°Chold
Purify the final library using according to Step 4.2 in Chromium Next GEM Single Cell ATAC Reagent Kits v1.1.

Sequencing preparation
Sequence on Illumina NovaSeq v1.5 platform with read setup : 36-8-48-36 (R1-I1-I2-R2) using custom sequencing primers:
Custom_primer_R1: GCGATCGAGGACGGCAGATGTGTATAAGAGACAG
Custom_primer_I2: CTGTCTCTTATACACATCTGCCGTCCTCGATCGC
R2 & I1 standard
Library structure:
P5 side:

P7 side:

We typically aim for ~25,000 read pairs per cell, meaning 125,000,000 reads for 5000 cells (~one 10x lane).
Desired outcome
Typical bioanalyzer trace of a successful experiment shows good and even distribution of fragment sizes and is not overtagmented or undertagmented. Both over- and under- tagmenation will lead to reduced complexity of the library.



