hyRAD (Suchan et al. 2016; Grealy et al.)
Alicia Grealy
Abstract
This bench protocol is based on the work of Tomasz Suchan, for performing hyRAD with RNA baits, with some changes.
Before start
Read Suchan et al. (2016)
Also see: dx.doi.org/10.17504/protocols.io.mt2c6qe by George Olah
Store MyOne C1 Streptavidin beads at 4 deg C in a fridge.
Steps
Probe synthesis
Preparation
"Suit up" in this order: hair net, nitrile gloves, facemask, coveralls, gumboots, booties, second pair of gloves.
Incubate in a thermal cycler at:
95 deg C for 1 min
Cool at a rate of 0.1 deg C / sec until the solution reaches 20 deg C
Store at -20 deg C.
Prepare the space by decontaminating surfaces with 10% household bleach followed by 70% ethanol. UV irradiate pipettes and racks. Racks should be bleached between subsequent uses and UV irradiated.
Ensure ice is available. Thaw reagents on ice as needed. Keep enzymes on ice at all times. Do not vortex enzymes to mix but mix by flicking the tube gently. Pulse centrifuge all reagents before opening.
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-bind Safelock tube | 1 | 10 mM Tris-HCl |
| 1.5 ml Lo-bind Safelock tube | 1 | 10X Annealing Buffer |
| 0.5 ml Lo-bind Safelock tube | 2 | P1 and P2 adapter oligos |
| 0.2 ml Lo-bind PCR tube | 1 | Annealing P1 and P2 adapter oligos |
Prepare all necessary buffers and UV decontaminate where appropriate.
| A | B | C | D |
|---|---|---|---|
| Buffer | Reagent | Volume to add | Final concentration in solution |
| 10 mM Tris-HCl | 1 M Tris-HCl | 10 ul | 10 mM |
| Ultrapure water | 990 ul | na | |
| 10 X Annealing Buffer | 1 M Tris-HCl | 100 ul | 100 mM |
| 5 M NaCl | 100 ul | 500 mM | |
| 0.5 M EDTA | 20 ul | 10 mM | |
| Ultrapure water | 780 ul | na |
Before resuspending oligos, pulse centrifuge to collect the pellet at the bottom of the tube. Add the appropriate buffer (see Materials) and vortex thoroughly. Store at -20 deg C. Dilute out the working concentrations (below) and store at -20 deg C when not in use. Thaw on ice. Vortex and pulse centrifuge after each thaw. Before beginning library preparation, make sure you have enough of each working stock prepared!
| A | B | C |
|---|---|---|
| Working stock | Reagent | Volume to add |
| 10 uM P5_Indexing_Primer | 100 uM Stock | 50 ul |
| Ultrapure water | 450 ul | |
| 10 uM P7_Indexing_Primer | 100 uM Stock | 50 ul |
| Ultrapure water | 450 ul |
Pre-program the thermal cycler.
Combine the following in a 0.2 ml Lo-bind PCR tube. Vortex and pulse centrifuge.
| A | B | C | D | E |
|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) |
| P1.1 adapter oligo (EcoRI) | 100 ul | 100 uM | 10 uM | 10 ul |
| P1.2 adapter oligo (EcoRI) | 100 ul | 100 uM | 10 uM | 10 ul |
| Annealing buffer | 100 ul | 10 X | 1 X | 10 ul |
| Ultrapure water | 100 ul | na | na | 70 ul |
Combine the following in a 0.2 ml Lo-bind PCR tube. Vortex and pulse centrifuge.
| A | B | C | D | E |
|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) |
| P2.1 adapter oligo (MspI) | 100 ul | 100 uM | 10 uM | 10 ul |
| P2.2 adapter oligo (MspI) | 100 ul | 100 uM | 10 uM | 10 ul |
| Annealing buffer | 100 ul | 10 X | 1 X | 10 ul |
| Ultrapure water | 100 ul | na | na | 70 ul |
Enzymatic digestion
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 2.2 Master mix |
| 0.2 ml Lo-Bind PCR tube | # of samples | Enzymatic digestion for samples |
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E |
|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) |
| CutSmart Buffer (NEB) | 50 ul | 10 X | 1 X | 5 ul |
| EcoRI-HF (NEB) | 50 ul | 20 U/ul | 0.4 U/ul (20 U) | 1 ul |
| MspI (NEB) | 50 ul | 20 U/ul | 0.4 U/ul (20 U) | 1 ul |
| Ultrapure water | 50 ul | na | na | 33 ul |
Add 40 ul of master mix to 10 ul of DNA (at 100 ng/ul ca. 1000 ng total) in a 0.2 ml Lo-bind PCR tube. Vortex and pulse centrifuge.
, though there is some degradation.")
Incubate in a thermal cycler at:
37 deg C for 4 hr
Hold at 4 deg C
Combine any replicates.
SPRI bead clean-up
Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 2X beads : reaction volume (i.e., 160 ul). Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 20 ul of 10 mM Tris-HCl.
Qubit
Measure the concentration using the Qubit HS kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Agarose gel electrophoresis
Electrophorese 2 ul of product (200-500 ng in 5-10 ul) on a 2% agarose gel.
2% Agarose Gel Electrophoresis

Adapter ligation
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 3.2 Master mix |
| 0.2 ml Lo-Bind PCR tube | # of samples x 2 | Reactions, two per sample |
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E |
|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) |
| CutSmart Buffer (NEB) | 20 ul | 10 X | 1 X | 2 ul |
| P1 adapter | 20 ul | 10 uM | 0.5 uM | 1 ul |
| P2 adapter | 20 ul | 20 uM | 1 uM | 1 ul |
| ATP | 20 ul | 100 mM | 1 mM | 0.2 ul |
| T4 DNA ligase | 20 ul | 400 U/ul | 20 U/ul (400 U) | 1 ul |
| Ultrapure water | 20 ul | na | na | 5.8 ul |
Add 11 ul of the above master mix to 9 ul of digested DNA in a 0.2 ml Lo-bind PCR tube. Vortex and pulse centrifuge.
Incubate in a thermal cycler at:
16 deg C for ca. 20 hr (16 hr - overnight)
Combine any replicates.
SPRI bead clean-up
Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 2X beads : reaction volume (i.e., 160 ul). Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 21 ul of 10 mM Tris-HCl.
Qubit
Measure the concentration using the Qubit HS kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
qPCR quant the RAD library
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 4.2 Master mix |
| 8-well strip qPCR tubes 0.1 ul profile | 1 | PCR amplification |
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 | C1 | C2 | V1 | x _____ rxn |
| Ultrapure water | 25 ul | na | na | 15.9 ul | |
| BSA | 25 ul | 10 mg /ml | 0.4 mg/ml | 1 ul | |
| ABI Gold PCR Buffer | 25 ul | 10 X | 1 X | 2.5 ul | |
| MgCl2 | 25 ul | 25 mM | 2.5 mM | 2.5 ul | |
| dNTPs | 25 ul | 25 mM | 0.25 mM | 0.25 ul | |
| ABI Taq Gold DNA polymerase | 25 ul | 5 U/ul | 0.05 U/ul | 0.25 ul | |
| SYBR Green | 25 ul | 5 X | 0.12 X | 0.6 ul | |
| IS7 | 25 ul | 10 uM | 0.2 uM | 0.5 ul | |
| IS8 | 25 ul | 10 uM | 0.2 uM | 0.5 ul |
Add 24 ul of master mix to the corresponding PCR tubes. Pulse centrifuge the tubes.
Dilute each library 1 in 20 (i.e., 1 ul of library in 19 ul of Ultrapure water). Vortex and pulse centrifuge.
Add 1 ul of DNA sample to the corresponding PCR tubes according to the scheme below. Pulse centrifuge the tubes.
e.g.,
| A | B | C | D |
|---|---|---|---|
| PCR NTC | |||
| PCR NTC | |||
| MD#033 Neat | |||
| MD#033 1 in 20 | |||
| MD#034 Neat | |||
| MD#034 1 in 20 | |||
Take the strip tubes to a post-PCR space. Place in thermal cycler and run the following program:
95 deg C for 10 min
Followed by 50 cycles of:
95 deg C for 30 sec
60 deg C for 30 sec
72 deg C for 30 sec
Agarose gel electrophoresis
Electrophorese 2 ul of product (200-500 ng in 5-10 ul) on a 2% agarose gel.
2% Agarose Gel Electrophoresis

Size selection using Pippin HT
Depending on the total yield and concentration determined using the Qubit (see Step 3.6 above), run approximately 1 ug of DNA in 20 ul (ca. 75 ng/ul) of each samples across a lane of a PippinHT electrophoresis system (2% gel, Marker 20B), selecting fragments 272 bp in size (272 bp peak with tight range 212-332 bp--this equates to an insert size of 180 bp) and following the manufacturer's instructions:
http://www.sagescience.com/wp-content/uploads/2015/10/PippinHT-Operations-Manual-Rev-B_460005.pdf
SPRI cleanup
Combine any replicates. Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 2X beads : reaction volume. Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 21 ul of Ultrapure water.
Qubit
Measure the concentration using the Qubit HS kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
qPCR quant the size-selected RAD library
Follow Steps 4.1 - 4.7 above to perform the qPCR if so desired.
Index / amplify the RAD library
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 7.2 Master mix |
| 8-well strip qPCR tubes 0.1 ul profile | 1 | PCR amplification |
Qubit
Measure the concentration using the Qubit HS kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Agarose gel electrophoresis
Electrophorese 5 ul of product (200-500 ng in 5-10 ul) on a 2% agarose gel.
2% Agarose Gel Electrophoresis
. This was not investigated further.")
Use a LabChip GXII or equivalent fragment analyser (HiSense kit) to measure the molarity of the libraries.
https://www.perkinelmer.com/Content/LST_Software_Downloads/LabChip_GX_User_Manual.pdf
Alternatively, use the Qubit concentration and the average fragment size seen on the gel to estimate the molarity of the libraries:
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge. Ensure to prepare enough master mix for multiple reactions per library plus pipetting error.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 | C1 | C2 | V1 | x _____ rxn |
| Ultrapure water | 50 ul | na | na | 29.5 ul | |
| KAPA High Fidelity Buffer | 50 ul | 5 X | 1 X | 10 ul | |
| P5_indexing_primer | 50 ul | 5 uM | 0.2 uM | 2 ul | Do not add to master mix |
| P7_indexing_primer | 50 ul | 5 uM | 0.2 uM | 2 ul | Do not add to master mix |
| KAPA HiFi Hot Start DNA Polymerase | 50 ul | 1 U/ul | 0.02 U/ul | 1 ul | |
| dNTPs | 50 ul | 25 uM | 0.25 uM | 0.5 ul |
Add 41 ul of master mix to the corresponding PCR tubes. Pulse centrifuge the tubes.
Add 2 ul of the corresponding forward indexing primer to the appropriate reaction tube. Pulse centrifuge the tubes.
Add 2 ul of the corresponding forward indexing primer to the appropriate reaction tube. Pulse centrifuge the tubes.
Add 5 ul of purified size-selected RAD library to the corresponding PCR tubes according to the scheme below. Pulse centrifuge the tubes.
e.g.,
| A | B | C | D |
|---|---|---|---|
| MD#033 | |||
| MD#033 | |||
| MD#033 | |||
| MD#033 | |||
| MD#034 | |||
| MD#034 | |||
| MD#034 | |||
| MD#034 |
Take the strip tubes to a post-PCR space. Place in thermal cycler and run the following program:
98 deg C for 10 min
Followed by 30 cycles of:
985 deg C for 20 sec
60 deg C for 30 sec
72 deg C for 40 sec
Then a final extension of:
72 deg C for 10 min
Pool replicate reactions into a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
SPRI bead clean-up
Purify the libraries using SeraMag Speed Beads or SeraMag Select using a1.4X beads : reaction volume. Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 40 ul of Ultrapure water.
Pool amplified libraries
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Pooled RAD library |
| 1.5 ml Lo-Bind Safelock tube | 1 | Aliquot of RAD library for sequencing |
Pool libraries in equimolar concentrations such that the total volume is is approximately 100 ul.
Aliquot 20 ul of the pooled RAD library into a new 1.5 ml Lo-bind Safelock tube for future sequencing. Store at -20 deg C. See Steps 58-63 in the following protocol to quantitate and sequence the final RAD library.
Adapter removal
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 9.2 master mix |
| 0.2 ml Lo-Bind PCR tube | 1 per 1 ug DNA | Adapter removal reaction |
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) | x ______ rxn |
| CutSmart Buffer (NEB) | 50 ul | 10 X | 1 X | 5 ul | |
| MspI | 50 ul | 20 U/ul | 0.4 U/ul (20 U) | 1 ul | |
| Ultrapure water | 50 ul | na | na | 25.4 ul |
Aliquot 31.4 ul of the above master mix into 0.2 ml Lo-bind PCR tubes.
Add 18.6 ul (1 ug) of pooled purified RAD library to each tube. Vortex and pulse centrifuge.
Incubate the reactions in a thermal cycler at:
37 deg C for 4 hr
Combine the replicates.
SPRI bead clean-up
Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 2X beads : reaction volume. Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 30 ul of Ultrapure water.
Qubit
Measure the concentration using the Qubit HS kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Agarose gel electrophoresis
Electrophorese 5 ul of product (200-500 ng in 5-10 ul) on a 2% agarose gel.
2% Agarose Gel Electrophoresis

In-vitro In-vitro transcription and biotinylation
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Step 10.3 master mix |
| 0.2 ml Lo-Bind PCR tube | # reactions | Transcription/biotinylation reaction |
| 0.2 ml Lo-Bind PCR tube | 2 | Aliquots of TURBO DNase and SuperaseIn RNAse inhibitor |
| 1.5 ml Lo-Bind Safelock tube | 1 | Combine probes for cleanup |
| RNeasy mini spin column | 1 | Purification |
| 1.5 ml Lo-Bind Safelock tube with the lid cut off | 1 | Elution |
| 1.5 ml Lo-Bind Safelock tube | 1 | Final tube for probes |
| 0.5 ml Lo-Bind Safelock tube | Depends on total ug of probes | 6-ul aliquots of probes |
Add 2.5 ul of SUPERase-IN RNAse Inhibitor (20 U/ul) to the eluate. Flick to mix and pulse centrifuge.
Qubit
Measure the concentration of the probes using the Qubit RNA kit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Fragment analyse
Examine the fragment length distribution of the probes using a fragment analyser such as the LabChip GXII RNA kit (or equivalent fragment analyser) following the manufacturer's instructions:
https://www.perkinelmer.com/Content/LST_Software_Downloads/LabChip_GX_User_Manual.pdf
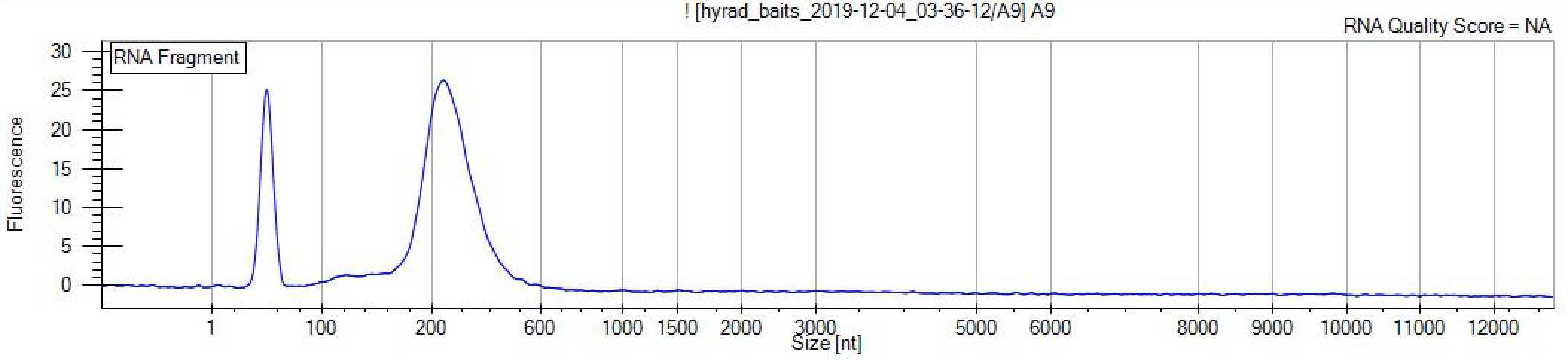
Dilute the RNA probes to 200 ng/ul with RNAse-free water. Aliquot into 6 ul batches in 0.5 ml Lo-bind Safelock tubes and store at -80 deg C.
Aliquot 10 ul of TURBO DNAse and 10 ul of SuperaseIn RNAse inhibitor into separate 0.2 ml Lo-bind tubes.
Combine the following in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) | x ______ rxn |
| Reaction Buffer (HiScribe kit) | 20 ul | 10 X | 0.75 X | 1.5 ul | |
| dATP | 20 ul | 100 uM | 7.5 uM | 1.5 ul | |
| dCTP | 20 ul | 100 uM | 7.5 uM | 1.5 ul | |
| dGTP | 20 ul | 100 uM | 7.5 uM | 1.5 ul | |
| dUTP | 20 ul | 100 uM | 5 uM | 1 ul | |
| biotin-UTP | 20 ul | 10 mM | 2.5 mM | 5 ul | |
| T7 RNA polymerase mix | 20 ul | ? | ? | 1.5 ul | |
| Ultrapure water | 20 ul | na | na | Adjust depending on the volume of library added |
Aliquot 13.5 ul of the above mater mix to 0.2 ml Lo-bind PCR tubes. Bring all tubes to a post-PCR space, including teh aliquots made in Step 10.2.
Concentrate the "probe set" (i.e., the pool RAD library with the adapters cut off) such that roughly 500 ng in 6.5 ul can be added per reaction using a SpeedVac, following the manufacturer's instructions:
Add 6.5 ul of the "probe set" (ca. 500 ng) to each reaction. Vortex and pulse centrifuge.
Incubate in a thermalcycler at:
37 deg C for 16 hr
In the last 30 min, add 2 ul of TURBO DNase (2 U/ul, or 4 U for up to 10 ug). Pipette up and down to mix.
Combine replicate reactions in a 1.5 ml Lo-bind tube. Top up to 100 ul with RNase-free water.
Cleanup using an RNeasy Mini Kit
Follow the manufacturer's instructions to purify the probes using an RNeasy Mini Kit:
http://www.bea.ki.se/documents/EN-RNeasy%20handbook.pdf
Elute in 60 ul RNase-free water.
Hybridisation
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 1.5 ml Lo-Bind Safelock tube | 1 | Mineral oil aliquot |
| 0.2 ml Lo-Bind PCR tube | # capture reactions x 2 | Hybridisation |
| 1.5 ml Lo-Bind Safelock tube | 2 | Step 14 and 15 master mixes |
Ensure all primer stocks are pepared in the correct buffer and that enough aliquots of the working concentrations are diluted out. Vortex and pulse centrifuge.
Thaw reagents. Flick all reagents to mix and pulse centrifuge where possible.
| A | B | C |
|---|---|---|
| Reagent | Stored at... | Thaw at... |
| 20 X SSPE | 4 deg C | Room temperature |
| 500 mM EDTA | 4 deg C | Room temperature |
| 50 X Denardt's Solution | -20 deg C | Room temperature |
| 10% SDS | 4 deg C | Room temperature |
| Chickent Cot-1 (HyBloc) | -20 deg C | On ice |
| FWD_blocking_primer | -20 deg C | On ice |
| REV_blocking_primer | -20 deg C | On ice |
| SUPERas-In RNAse inhibitor | -20 deg C | On ice |
| Salmon Sperm DNA | -20 deg C | On ice |
Combine the following " BLOCKS " master mix in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) | x ______ rxn |
| Chicken Cot-1 (HyBloc) | 3.25 ul | 1 ug/ul | 0.77 ug/ul | 2.5 ul | |
| Salmon Sperm DNA | 3.25 ul | 10 ug/ul | 0.77 ug/ul | 0.25 ul | |
| FWD_blocking_primer | 3.25 ul | 200 uM | 15.4 uM | 0.25 ul | |
| REV_blockg_primer | 3.25 ul | 200 uM | 15.4 uM | 0.25 ul |
"BLOCKS" master mix
Aliquot 3 ul of the BLOCKS master mix into 0.2 ml Lo-bind tubes, one for each capture reaction.
Combine the following " HYBS " master mix in a 1.5 ml Lo-bind Safelock tube. Vortex and pulse centrifuge.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 (reaction volume) | C1 (stock concentration) | C2 (concentration in reaction) | V1 (volume to add) | x ______ rxn |
| SSPE | 20 ul | 20 X | 9 X | 9 ul | |
| EDTA | 20 ul | 0.5 M | 0.0125 M | 0.5 ul | |
| Denhardt's solution | 20 ul | 50 X | 8.75 X | 3.5 ul | |
| SDS | 20 ul | 10 % | 0.25 % | 0.5 ul | |
| SUPERase-In RNase inhibitor | 20 ul | 20 U/ul | 1 U/ul | 1 ul |
"HYBS" master mix
Aliquot 14.5 ul of the HYBS master mix into 0.2 ml Lo-bind tubes, one for each capture reaction.
Bring all the tubes to the post-PCR space.
Pre-program the thermal cycler:
95 deg C for 5 min
60 deg C for 5 min
60 deg C for hold
Thaw a 6-ul aliquot of the probes from Step 10.13 on ice.
Add 5.5 ul of baits (ca. 500-1000 ng at 100-200 ng/ul) to each HYBS reaction. Flick to mix and pulse centrifuge. Place on ice.
Add 7 ul (100-500 ng) of shotgun library to the corresponding BLOCKS tube. Flick to mix and pulse centrifuge.
Transfer the BLOCKS tubes to a thermalcycler and start the program. Allow it to proceed through Step 1 (i.e., 95 deg C for 5 min).
When the thermalcycler reaches Step 2, transfer the HYBS tubes to the thermalcyler. Allow it to proceed through Step 2 (i.e., 60 deg C for 5 min).
When the thermalcycler reaches Step 3, transfer 18 ul from the HYBS tubes to the corresponding BLOCKS tubes. Pipette to mix. Discard the HYBS tubes.
Add 15 ul of mineral oil to the top of each reaction.
Allow the thermalcycler to proceed through Step 3 for 42 hr (i.e., 60 deg C for 42 hr).
Enrichment
Label tubes.
| A | B | C |
|---|---|---|
| Tube | Qty | For ... |
| 50 ml Falcon tubes | 3 | Wash buffer 2, Wash buffer 2.2, Binding buffer |
| 15 ml Falcon tubes | 3 | Aliquots for Wash buffer 2.2 and Binding buffer, and 10 mM Tris-HCl/0.05% Tween-20 |
| 1.5 ml Lo-bind Safelock tube | 1 | Aliquot for MyOne C1 Streptavidin beads |
| 1.5 ml Lo-bind Safelock tube | 3 x # hybridisation reactions | Enrichment |
Prepare all necessary buffers and UV decontaminate.
| A | B | C | D |
|---|---|---|---|
| Buffer | Reagent | Volume | C2 |
| Wash buffer 2 | 20 X SSC | 100 ul | 0.1 X |
| 10% SDS | 200 ul | 0.1 % | |
| Ultrapure water | 19.7 ml | na | |
| Wash buffer 2.2 | 10% SDS | 400 ul | 0.08% |
| Wash buffer 2 | 10 ml | na | |
| Ultrapure water | 39.6 ml | na | |
| Binding buffer | 5 M NaCl | 10 ml | 1 M |
| 1 M Tris-HCl | 500 ul | 10 mM | |
| 0.5 M EDTA | 100 ul | 1 mM | |
| Ultrapure water | 39.4 ml | na | |
| 10 mM Tris-HCl, 0.05% Tween-20 | 1 M Tris-HCl | 500 ul | 10 mM |
| 100% Tween-20 | 25 ul | 0.05% | |
| Ultrapure water | 49.475 ml | na |
Aliquot reagents for to take to the post-PCR space.
| A | B |
|---|---|
| 700 ul * # reactions | Binding buffer |
| 30 ul * # reactions | MyOne C1 Streptavidin beads |
| 35 ul * # reactions | 10 mM Tris-HCl/0.05% Tween-20 |
| 1600 ul * # reactions | Wash Buffer 2.2 |
Set a water bath or thermalshaker to 55 deg C. Warm Wash buffer 2.2 to 55 deg C for 45 min.
Set a heat block to 95 deg C.
Pellet the MyOne C1 Streptavidin beads for 2 min with the magnetic rack. Discard the supernatant.
Add 200 ul * # reactions of Binding buffer to the beads. Vortex and pulse centrifuge.
Pellet the beads with the magnetic rack and discard the supernatant.
Repeat Step 34-35 two more times for a total of 3 washes.
Resuspend the beads in 70 ul * # reactions of Binding buffer.
Aliquot 70 ul of beads into a 1.5 ml Lo-bind Eppendorf tube (1 per reaction).
Warm the bead aliquots to 55 deg C in the thermoshaker/water bath for 2 minutes.
Transfer the hybridised libraries at 60 deg C to the bead aliquots. Pipette to mix.
Incubate the hybridised libraries and beads in the thermoshaker/waterbath for 30 min at 55 deg C. Agitate every 5 minutes by flicking or gently continuously shake.
Pulse centrifuge. Pellet the beads with the magnetic rack. Discard the supernatant.
Add 500 ul heated Wash Buffer 2.2 to the beads. Vortex and pulse centrifuge.
Incubate 10 min at 55 deg C in the thermoshaker/water bath. Agitate every 2 min by flicking.
Pulse centrifuge. Pellet the beads with the magnetic rack. Discard the supernatant.
Repeat the wash steps above (Steps 43-45) two more times for a total of 3 washes.
Add 30 ul of 10 mM Tris-HCl/0.05% Tween-20 to the washed beads. Resuspend by pipetting.
Incubate at 95 deg C in a heat block for 5 min.
Pellet the beads with a magnetic rack and transfer the supernatant to a clean 1.5 ml tube.
At this point you can treat with RNAseA to remove any RNA or put RNAse A in the PCR but it is not necessary as the RNA will not amplify.
Dilute the captured libraries 1 in 10 in Ultrapure water (i.e., 1 ul in 9 ul Ultrapure water). Vortex and pulse centrifuge.
Library amplification
Label tubes.
Thaw reagents on ice.
Ensure that all primer stocks are prepared in the correct buffer and that enough aliquots of the working concentrations are diluted out. Vortex and pulse centrifuge.
| A | B | C |
|---|---|---|
| Oligo | Reagent | Volume |
| 10 uM P5_primer | 100 uM P5 | 50 ul |
| Ultrapure water | 450 ul | |
| 10 uM P7_primer | 100 uM P7 | 50 ul |
| Ultrapure water | 450 ul |
Make up the following master mix in a 1.5 ml Lo-bind tube.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 | C1 | C2 | V1 | x _____ rxn |
| Ultrapure water | 25 ul | na | na | 10.9 | |
| BSA | 25 ul | 10 mg /ml | 0.4 mg/ml | 1 ul | |
| ABI Gold PCR Buffer | 25 ul | 10 X | 1 X | 2.5 ul | |
| MgCl2 | 25 ul | 25 mM | 2.5 mM | 2.5 ul | |
| dNTPs | 25 ul | 25 mM | 0.25 mM | 0.25 ul | |
| ABI Taq Gold DNA polymerase | 25 ul | 5 U/ul | 0.05 U/ul | 0.25 ul | |
| SYBR Green | 25 ul | 5 X | 0.12 X | 0.6 ul | |
| P5 | 25 ul | 10 uM | 0.4 uM | 1 ul | |
| P7 | 25 ul | 10 uM | 0.4 uM | 1 ul |
Master mix for end-point PCR
Add 22.5 ul of master mix to each PCR tube following the schematic below. Pulse centrifuge the tubes.
| A | B |
|---|---|
| Capture001 Neat | |
| Capture001 1in10 | |
| Capture002 Neat | |
| Capture002 1in10 | |
| Capture003 Neat | |
| Capture003 1in10 | |
| PCR NTC | |
| PCR NTC |
Make up the following master mix in a 1.5 ml Lo-bind tube.
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Reagent | V2 | C1 | C2 | V1 | x _____ rxn |
| Ultrapure water | 25 ul | na | na | 10.9 | |
| BSA | 25 ul | 10 mg /ml | 0.4 mg/ml | 1 ul | |
| ABI Gold PCR Buffer | 25 ul | 10 X | 1 X | 2.5 ul | |
| MgCl2 | 25 ul | 25 mM | 2.5 mM | 2.5 ul | |
| dNTPs | 25 ul | 25 mM | 0.25 mM | 0.25 ul | |
| ABI Taq Gold DNA polymerase | 25 ul | 5 U/ul | 0.05 U/ul | 0.25 ul | |
| SYBR Green | 25 ul | 5 X | 0.12 X | 0.6 ul | |
| P5 | 25 ul | 10 uM | 0.4 uM | 1 ul | |
| P7 | 25 ul | 10 uM | 0.4 uM | 1 ul |
Master mix for final PCR
Add 20 ul of master mix to each PCR tube following the schematic below. Pulse centrifuge the tubes.
| A | B |
|---|---|
| Capture001 Neat | Capture003 Neat |
| Capture001 Neat | Capture003 Neat |
| Capture001 Neat | Capture003 Neat |
| Capture001 Neat | Capture003 Neat |
| Capture002 Neat | |
| Capture002 Neat | |
| Capture002 Neat | |
| Capture002 Neat |
Pulse centrifuge the 8-well qPCR strip tubes containing the master mix. Bring to the post-PCR space.
To the end-point PCR, add 2.5 ul of both neat and 1in10 captured library to the corresponding tubes. Add 2.5 ul nuclease free water to the remaining wells as PCR no-template controls. Vortex and pulse centrifuge.
Place tubes in the thermal cycler and run the following PCR program:
95 deg C for 10 min
Followed by 50 cycles of:
95 deg C for 30 sec
60 deg C for 30 sec
72 deg C for 30 sec
When the PCR is finished, determine the optimal number of cycles to give in the final PCR to ensure libraries are not over-amplified. i.e., stop the PCR during the linear phase of amplification.
Agarose gel electrophoresis
Run 10 ul of PCR product from the first PCR on a 2% agarose gel electrophoresis.
To the second PCR add 5 ul of the neat library (as long as amplification was NOT inhibited in the first PCR; if amplification efficiency was poor, amplify a dilution and do more replicates). Perform enough replicates to amplify the entire captured library.
Perform Step 61 above, but stop the PCR during the linear phase of amplification as determined by the end-point PCR above.
Combine replicates in a clean 1.5 mL Lo-bind Safelcok tube. Vortex and pulse centrifuge.
Purify
Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 1.6X beads : reaction volume (i.e., 160 ul). Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 40 ul of Ultrapure water.
Size select enriched libraries
Run each enriched library in duplicate across two lanes (20 ul each) of a PippinHT electrophoresis system (2% gel, Marker 20B), selecting fragments between 160-500 bp and following the manufacturer's instructions:
http://www.sagescience.com/wp-content/uploads/2015/10/PippinHT-Operations-Manual-Rev-B_460005.pdf
Purify
Combine replicates. Purify the libraries using SeraMag Speed Beads or SeraMag Select using a 2X beads : reaction volume (i.e., 160 ul). Follow the guidelines below:
https://www.gelifesciences.co.jp/catalog/pdf/SeraMagSelect_UserGuide.pdf
Elute in 25 ul of Ultrapure water.
Pool enriched libraries
Qubit
Measure the concentration of the neat library and these dilutions in duplicate on the Qubit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Dilute the libraries to 5 ng/ul in Ultrapure water in a total volume of 5-10 ul.
Fragment analyse
Use a LabChip GXII or equivalent fragment analyser (HiSense kit) to measure the molarity of the libraries between 160-500 bp.
https://www.perkinelmer.com/Content/LST_Software_Downloads/LabChip_GX_User_Manual.pdf
Pool enriched libraries in equimolar concentrations.
Quantitate the final sequencing library
Dilute the libraries 1/2, 1/5, 1/10 in Ultrapure water (i.e., create a serial dilution in 10 ul volume).
Qubit
Measure the concentration of the neat library and these dilutions in duplicate on the Qubit following the manufacturer's instructions.
https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf
Fragment analyse
Measure the molarity of the neat library and dilutions on a LabChip GXII Hisense kit (or equivalent fragment analyser) following the manufacturer's instructions:
https://www.perkinelmer.com/Content/LST_Software_Downloads/LabChip_GX_User_Manual.pdf
Based on the average fragment length and Qubit measurement, calculate the molarity of the library dilutions. Create a standard curve to check that the concentrations are linear. If they can be "trusted", extrapolate the neat concentration based on the dilutions. Average all the measurements of the neat concentration to get the best estimate of the library molarity.
Sequence
Dilute the library to between 2-4 nM in Ultrapure water.
Follow the manufacturer's instructions to perform the sequencnig run on your Illumina platform of choice.
