A fluorescence-based in vitro scrambling assay for yeast MCP1 and human XK
Zhouping Hong, Karin Reinisch
Disclaimer
DISCLAIMER – FOR INFORMATIONAL PURPOSES ONLY; USE AT YOUR OWN RISK
The protocol content here is for informational purposes only and does not constitute legal, medical, clinical, or safety advice, or otherwise; content added to protocols.io is not peer reviewed and may not have undergone a formal approval of any kind. Information presented in this protocol should not substitute for independent professional judgment, advice, diagnosis, or treatment. Any action you take or refrain from taking using or relying upon the information presented here is strictly at your own risk. You agree that neither the Company nor any of the authors, contributors, administrators, or anyone else associated with protocols.io, can be held responsible for your use of the information contained in or linked to this protocol or any of our Sites/Apps and Services.
Abstract
VPS13 proteins are proposed to function at contact sites between organelles as bridges for lipids to move directionally and in bulk between organellar membranes. VPS13s are found to interact with integral membrane proteins, like MCP1 in yeast or XK in humans. We showed that MCP1 and XK scramble phospholipids in vitro. Here I describe the detailed procedure of purification, reconstitution, and scrambling assay for both MCP1 and XK.
Steps
Protein purification
Constructs encoding MCP1 (pCMV10-3xFLAG-Prs-MCP1) or XK (pCMV10-3xFLAG-Prs-XK) were transfected into Expi293 cells at the density of ~3 million/ml. The protein expression was enhanced with 3.5millimolar (mM) valproic acid (VPA) 18 hours after transfection. The cells were harvested 48 hours after transfection and cell pellets were stored at -80°C.
MCP1 purification
The cell pellets were resuspended in buffer A (50 mM HEPES, pH7.0, 500 mM NaCl, 1 mM TCEP, 10% glycerol) containing 1×
To solubilize the protein, powdered DDM was added to the lysate at a final concentration of 1% w/v, and the lysate was gently agitated in the cold room for 90 minutes. Cell lysates were clarified via centrifugation at 100,000 g for 60 minutes and the supernatant was incubated with anti-FLAG M2 resin (Sigma Aldrich), which was pre-equilibrated with buffer B (buffer A, 0.02% DDM), at 4°C for 2 hours, and then the resin was washed with buffer B. To remove the chaperone, the resin was incubated with buffer B containing 5 mM MgCl2, 2.5 mM ATP containing buffer C at 4 °C overnight.
The next day, bound proteins were further washed with buffer B, then eluted using 0.25mg/ml 3xFlag peptide in Buffer B. The proteins were concentrated in a 10-kD molecular weight cutoff (MWCO) Amicon centrifugal filtration device (UFC501024) and quantified by Coomassie blue staining using BSA standards.
XK purification
The cell pellets were resuspended in buffer C (50 mM HEPES, pH 7.8, 200 mM NaCl, 1 mM TCEP, 10% Glycerol) containing 1× cOmplete EDTA-free protease inhibitor cocktail (Roche) and lysed using a Dounce homogenizer (15~20 passes).
To solubilize the protein, powdered GDN was added to the lysate at a final concentration of 1.5 % w/v, and the lysate was gently agitated in the cold room for 90 minutes. Cell lysates were clarified via centrifugation at 100,000 g for 60 minutes and the supernatant was incubated with anti-FLAG M2 resin (Sigma Aldrich), which was pre-equilibrated with buffer D (buffer C, 0.02% GDN), at 4°C for 2 hours, and then the resin was washed with buffer D. To remove the chaperone, the resin was incubated with buffer D containing 5 mM MgCl2, 2.5 mM ATP containing buffer C at 4 °C overnight.
The next day, bound proteins were further washed with buffer D, then eluted using 0.25mg/ml 3xFlag peptide in Buffer D. The protein was gel filtrated with Superdex 200 increase 10/300 column (Cytiva) and concentrated in a 10-kD molecular weight cutoff (MWCO) Amicon centrifugal filtration device (UFC501024) and quantified by Coomassie blue staining using BSA standards.
Liposome preparation
90% POPC37°C for 60 minutes, with vortexing every 10 minutes, and then freeze-thawed for ten cycles. Liposomes were extruded 31 times through a 400 nm polycarbonate filter and used within 6 hours.
Proteoliposome preparation
Determine the optimal swelling conditions by a swelling titration assay
Wash the cuvette with miniQ water and buffer E or F.
Blank: set the parameter to OD540, set the blank point with your reconstitution buffer (buffer E or F).
Dry the cuvette completely with an air duster.
Add 800μl liposome solution to the cuvette, record the OD540. If the readout is too high, like 2.0, dilute the liposome solution and read again, until the original read is ~1.
Add 1μl of 10% Triton X-100 each time, and cap the cuvette, invert to mix for 30 seconds and record the OD540. repeat until the liposomes are completely dissolved.
Make a curve of the swelling titration assay. The typical curve with Triton X-100 looks like this, use the Triton X-100 concentration between saturation point and dissolving point (close to dissolving point, see curve below).
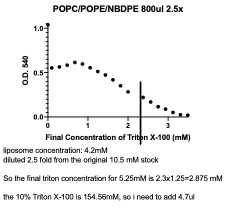
Prepare proteoliposomes: For each reaction, we have 250μl volume in total, the protein volume shouldn’t be more than 10%, otherwise, the detergent in the protein sample could interfere with the reconstitution. You can have 125 μl liposomes in each reaction so the final concentration of lipids is 5.25mM, and 25μl protein sample, and the corresponding Triton X-100 amount (4.7μl), and fill the volume with your reconstitution buffer.
Swelling the liposomes: add the desired amount of Triton X-100 to 125μl liposomes, and add reconstitution buffer, agitate at room temperature for 2 hours. (add NBD-glucose to the NBD-glucose control group at this step)
Add the pre-purified protein into the solution, agitate for 1h at room temperature.
Prepare BioBeads: take some Biobeads into a 50ml tube, wash with methanol 3 times, miniQ water 2 times, reconstitution buffer once. For each wash, add liquid to the BioBeads to fill the tube, agitate at room temperature for 10min, discard the liquid. Pour the BioBeads to KimWIpe to dry it.
Detergent removal: add ¼ of BioBeads to the solution, agitate at room temperature for 1h; add another ¼ of BioBeads to the solution, agitate at room temperature for 2h; transfer the solution to a new tube, add the last ½ of BioBeads, agitate at 4°C overnight.
The next day, transfer the solution into a new 1.5ml tube.
Determine the reconstitution efficiency: use
Prepare different concentrations of OptiPrep using the reconstitution buffer. Each gradient needs at least 1ml. These will be the gradient solution
Mix your sample with equal-volume 40% OptiPrep to get the 20% concentration gradient solution
Add the gradient solution into the ultracentrifuge tube: lot No: Z20214SCA (750μl)
300 μl 20%; 200μl of the other gradients.
Use gel loading tips, slowly pipette down the solution to prevent disturbing the gradient.
Ultracentrifuge at 45,000 rpm 4°C for 1~1.5 hours, using SW 55 rotor. The protein itself goes to the bottom of the tube, and the proteoliposomes are at the very top. Separate the solution from the top, middle and bottom, run them on the SDS-PAGE gel.


Scrambling assay
The scrambling assay was performed at 30°C in 96-well plates, with 100 μl reaction volumes of liposomes/proteoliposomes (~260 μM final lipid concentration) prepared as described above. To assess scrambling, NBD fluorescence after addition of dithionite (to 5 mM) was monitored (excitation at 460 nm, emission at 538 nm) using the Synergy H1 Hybrid Multi-Mode Reader (BioTek). Finally, additional dithionite (5 mM) and Triton X-100 (0.5%) were added. The Triton X-100 dissolves the liposomes, allowing complete quenching of the NBD.
Set the Synergy H1 plate reader temperature as 30°C
Add 95μl of the reconstitution buffer, 5μl of liposome/proteoliposome sample. Mix well.
Monitor the NBD fluorescence with excitation at 460 nm, emission at 538 nm for 5 minutes.
Prepare 100mM dithionite with 50mM Tris-HCl, pH 8.0 solution.
Add 5μl 100mM dithionite to each well, mix a little bit, monitor the emission changes for 30 minutes or 10 minutes.
Prepare another aliquot of dithionite. Add 5μl 10% Triton X-100 and 5μl 100mM dithionite to each well, mix a little bit. Monitor the changes for 5min, it should go to zero directly.
A similar protocol was used for the NBD-glucose leakiness control assay (Goren et al., 2014; Ploier and Menon, 2016), except that no NBD-lipids were incorporated into the liposomes or proteoliposomes. Instead, NBD-glucose (3 mM) was added during the destabilization step.
